Research
As synthetic organic chemists work toward more sustainable approaches to complex transformations, preparative electrochemical methods become natural choices due to their high product efficiency, mild conditions, safety, and ease of scalability. Despite these benefits, most practicing synthetic organic chemists lack the tools and methods to develop or exploit electrochemical techniques. This center aims to combine the expertise of electrochemists with modern synthetic organic chemists to provide the knowledge base, techniques, and instrumentation that will enable widespread adoption of preparative electrochemical methods by both laboratory and industrial scale organic chemists.
The Center is focused specifically on three main thrusts: utilizing electrochemical methods for C-H functionalization, utilizing electrochemical approaches to traditional cross-coupling reactions, and arene functionalization via mediated anodic oxidation.
Mechanistic Studies into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization
Christopher Sandford, Lydia R. Fries, Tyler E. Ball, Shelley D. Minteer, and Matthew S Sigman
Mechanistic Studies into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization
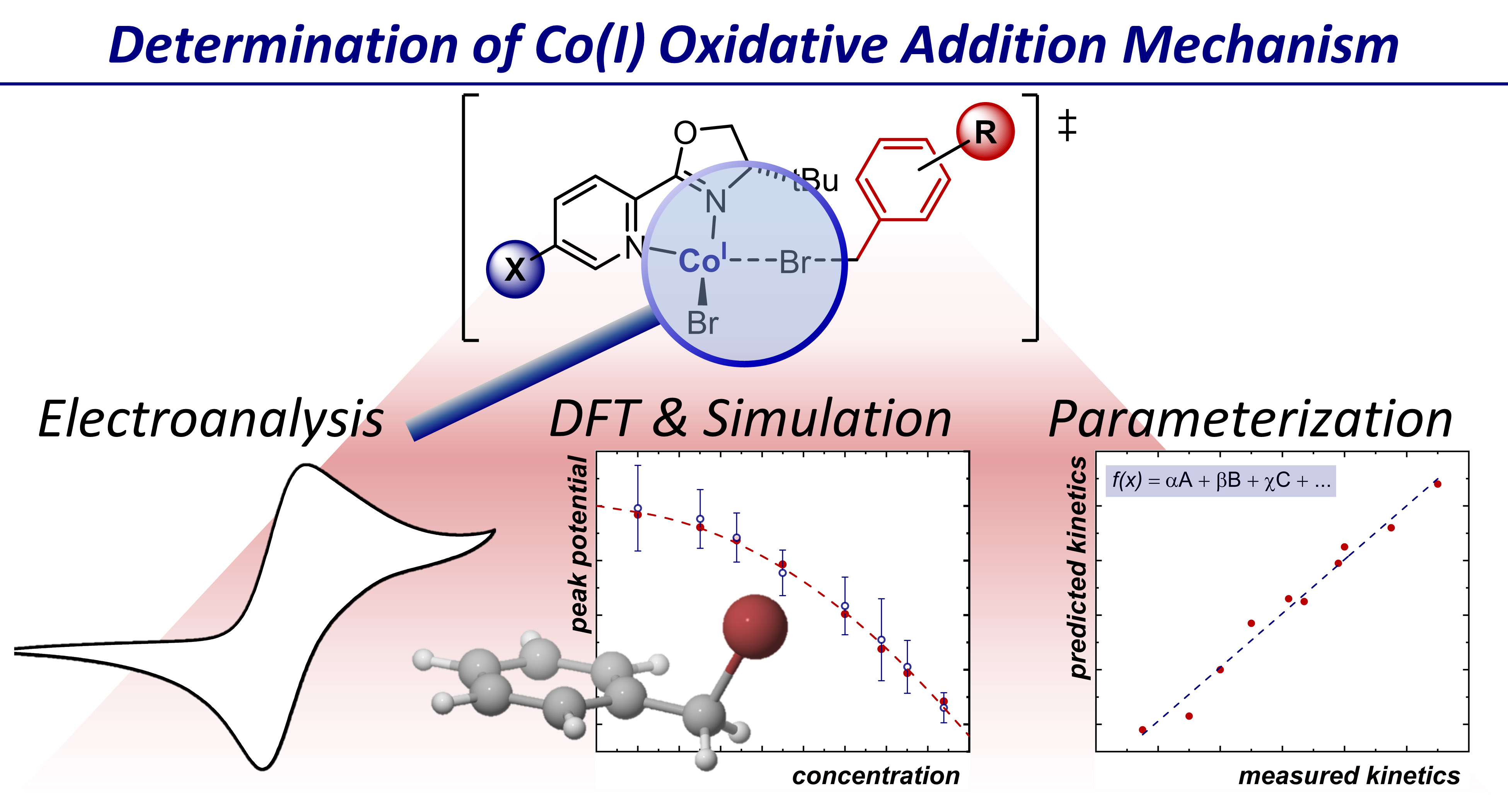
The oxidative addition of organic electrophiles into electrochemically generated Co(I) complexes has been widely utilized as a strategy to produce carbon-centered radicals when cobalt is ligated by a polydentate ligand. Changing to a bidentate ligand provides the opportunity to access discrete Co(III)ñC bonded complexes for alternative reactivity, but knowledge of how ligand and/or substrate structures affect catalytic steps is pivotal to reaction design and catalyst optimization. In this vein, experimental studies which can determine the exact nature of elementary organometallic steps remain limited, especially for single-electron oxidative addition pathways. Herein, we utilize cyclic voltammetry combined with simulations to obtain kinetic and thermodynamic properties of the two-step, halogen-atom abstraction mechanism, validated by analyzing kinetic isotope and substituent effects. Complex Hammett relationships could be disentangled to allow understanding of individual effects on activation energy barriers and equilibrium constants, and DFT-derived parameters used to build predictive statistical models for rates of new ligand/substrate combinations. DOI: 10.1021/jacs.9b10771