Publications
An Alternative Mechanistic Paradigm for the Electrochemical C-Terminal Decarboxylation of Peptides
ChemElectroChem
Adam J. Sowers, Kevin D. Moeller, Kim S. Halskov
An Alternative Mechanistic Paradigm for the Electrochemical C-Terminal Decarboxylation of Peptides
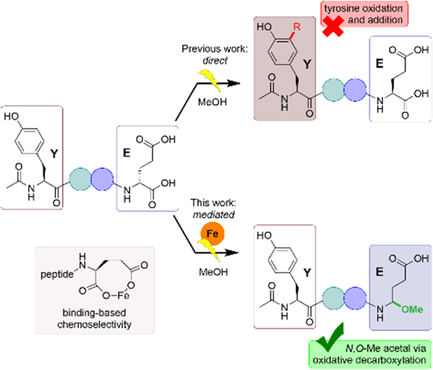
The C-terminal decarboxylation of peptides provides an important opportunity to synthesize modern peptide pharmaceuticals that contain C-terminal amides. This transformation can be achieved by electrochemical oxidation; however, the standard implementation depends on oxidation potential for selectivity which may represent a challenge when amino acid residues containing electroactive side chains are present. To address this limitation, an alternative mechanistic paradigm has been introduced for selective decarboxylation of a C-terminal carboxylate, one that relies on a chelation event. In a proof-of-principle experiment used to probe and define the viability of this mechanism, it is demonstrated that the combination of an iron mediator and a C-terminal glutamate residue can be used to conduct the reaction in the presence of the more electron-rich tyrosine residue frequently found in medicinally active peptides. Investigations into the reaction specifics and the scope/limitations provide key insights into the reaction mechanism and how such processes can be optimized. The success of the method highlighted here points to a more general binding-based approach to drive C-terminal decarboxylation that utilizes a functional group motif not possible at any other position in a peptide.
Ni-Catalyzed Asymmetric Reductive Arylation of α-Substituted Imides
JACS
Li-Ming Chen, Chungkeun Shin, Travis J. DeLano, Alba Carretero-Cerdán, Golsa Gheibi, Sarah E. Reisman
Ni-Catalyzed Asymmetric Reductive Arylation of α-Substituted Imides
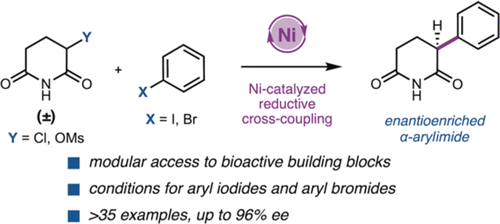
α-Aryl imides are common structural motifs in bioactive molecules and proteolysis-targeting chimeras designed for targeted protein degradation. An asymmetric Ni-catalyzed reductive cross-coupling of imide electrophiles and (hetero)aryl halides has been developed to synthesize enantioenriched α-arylglutarimides from simple starting materials. Judicious selection of electrophile pairs allows for coupling of both electron-rich and electron-deficient (hetero)aryl halides in good yields and enantioselectivities.
Intentional Formation of Persistent Surface Redox Mediators by Adsorption of Polyconjugated Carbonyl Complexes to Pd Nanoparticles
JACS
Jason S. Adams, Mayank Tanwar, Haoyu Chen, Sucharita Vijayaraghavan, Tomas Ricciardulli, Matthew Neurock, David W. Flaherty
Intentional Formation of Persistent Surface Redox Mediators by Adsorption of Polyconjugated Carbonyl Complexes to Pd Nanoparticles
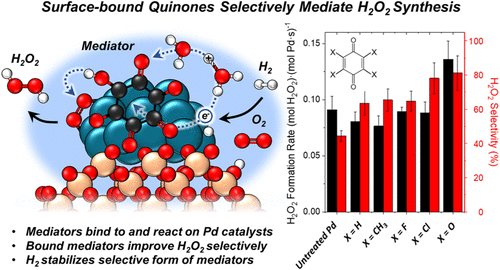
Adsorbing polyconjugated carbonyl and aromatic species to Pd nanoparticles forms persistent intermediates that mediate reactions between hydrogen and oxygen-derived species. These surface redox mediators form in situ and increase selectivities toward H2O2 formation (∼65–85%) compared to unmodified Pd nanoparticles (∼45%). Infrared spectroscopy, temperature-programmed oxidation measurements, and ab initio calculations show that these species adsorb irreversibly to Pd surfaces and persist over extended periods of catalysis. Combined rates and kinetic isotope effect measurements and simulations suggest that carbonyl groups of bound organics react heterolytically with hydrogen to form partially hydrogenated oxygenated complexes. Subsequently, these organic species transfer proton–electron pairs to O2-derived surface species via pathways that favor H2O2 over H2O formation on Pd nanoparticles. Computational and experimental measurements show redox pathways mediated by partially hydrogenated carbonyl species form H2O2 with lower barriers than competing processes while also obstructing O–O bond dissociation during H2O formation. For example, adsorption and hydrogenation of hexaketocyclohexane on Pd forms species that react with oxygen with high H2O2 selectivities (85 ± 8%) for 130 h on stream in flowing water without additional promoters or cosolvents. These paths resemble the anthraquinone auto-oxidation process (AAOP) used for industrial H2O2 production. These surface-bound species form partially hydrogenated intermediates that mediate H2O2 formation with high rates and selectivities, comparable to AAOP but on a single catalytic nanoparticle in pure water without organic solvents or multiunit reaction-separation chains. The molecular insights developed herein provide strategies to avoid organic solvents in selective processes and circumvent their associated process costs and environmental impacts.
Reduction by Oxidation: Selective Hydrodehalogenation of Aryl Halides by Mediated Oxalate Oxidation
JACS
Joshua A. Beeler, Rune P. Walkingshaw, Safiya A.S. Hamud, Henry S. White
Reduction by Oxidation: Selective Hydrodehalogenation of Aryl Halides by Mediated Oxalate Oxidation
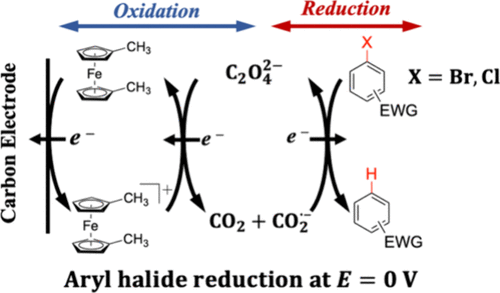
Electro-organic reduction reactions are canonically carried out at a cathode at which a significant negative potential is applied. Specifically, at carbon electrodes, aryl bromides and chlorides undergo heterogeneous reduction in organic solvents at potentials more negative than −2 V vs E0′ for the Fc/Fc+ couple (Fc = ferrocene). To decrease the overpotential for reduction reactions, homogeneous or heterogeneous electrocatalysis strategies are often employed. Here, we present an electrochemical method to reduce aryl bromides and chlorides that is initiated by an oxidation reaction at very mild potentials (∼0 V vs Fc/Fc+). Specifically, electrochemical oxidation of an outer-sphere redox mediator, 1,1-dimethylferrocene, in dry N,N-dimethylformamide (DMF) containing oxalate (C2O42–), results in the homogeneous one-electron oxidation of C2O42–. The resulting C2O4•– decomposes in ∼1 μs to release the carbon dioxide radical anion (CO2•–), a potent reductant that is oxidized to CO2 at –2.68 V vs Fc/Fc+. In this way, an oxidation reaction at low electrode potentials enables homogeneous reduction of aryl bromides and chlorides, which are otherwise directly reduced at very negative potentials. Using this method, selective hydrodehalogenations of electron-deficient aryl bromides and chlorides are carried out at a reticulated vitreous carbon anode with up to quantitative conversion yields. Cyclic voltammetry and finite difference simulations are used to characterize the hydrodehalogenation of 4-bromobenzonitrile via C2O42– oxidation. Additionally, we show that the efficiency of hydrodehalogenation is tuned by deliberate additions of water to DMF solutions, leading to a substantial improvement in overall conversion yields without interference from water or proton reduction.
Enhancing the Selectivity and Confinement of the Cu(II)-Mediated Chan-Lam Coupling for Use in Building Point-of-Care Diagnostics
ChemElectroChem
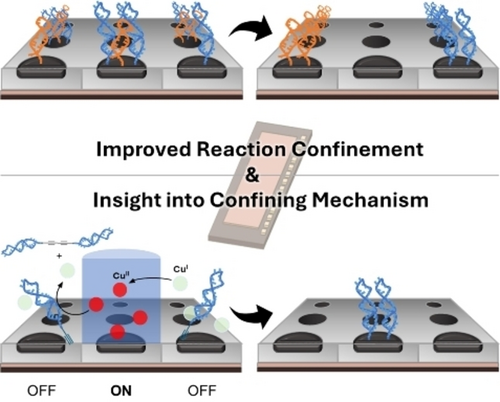
Yu-Chia Chang, Kevin D. Moeller
Enhancing the Selectivity and Confinement of the Cu(II)-Mediated Chan-Lam Coupling for Use in Building Point-of-Care Diagnostics
The Cu(II)-mediated Chan-Lam coupling reaction offers several benefits for developing point-of-care detection devices on microelectrode arrays. However, achieving selectivity on borate ester-based polymer surfaces has proven difficult due to background reactions. Fluorescence-based studies were conducted using fluorescently labeled acetylene nucleophiles. Initial experiments revealed significant background fluorescence across the electrode array, indicating selectivity issues. Further investigation uncovered significant background reactions occurring even without copper. To address this, a strategy utilizing an arylbromide-based polymer was developed, enhancing reaction selectivity by minimizing background non-specific reactions. Exploration into the confinement mechanism revealed the role of acetylene in forming dimers, facilitating rapid consumption of Cu(II) reagents that escaped from the specific electrodes used. These findings offer a way to construct devices for the multiplex point-of-care detection of metabolites, improving performance and accuracy in diagnostic devices.
Data-Driven Workflow for the Development and Discovery of N-Oxyl Hydrogen Atom Transfer Catalysts
ACS Central Science
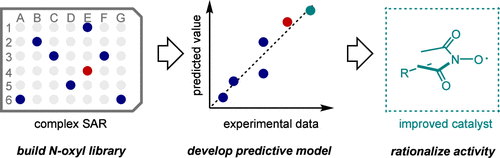
Cheng Yang, Thérèse Wild, Yulia Rakova, Stephen Maldonado, Matthew S. Sigman, Corey R. J. Stephenson
Data-Driven Workflow for the Development and Discovery of N-Oxyl Hydrogen Atom Transfer Catalysts
N-oxyl species are promising hydrogen atom transfer (HAT) catalysts to advance C–H bond activation reactions. However, because of the complex structure–activity relationship within the N-oxyl structure, catalyst optimization is a key challenge, particularly for simultaneous improvement across multiple parameters. This paper describes a data-driven approach to optimize N-oxyl hydrogen atom transfer catalysts. A focused library of 50 N-hydroxy compounds was synthesized and characterized by three parameters─oxidation peak potential, HAT reactivity, and stability─to generate a database. Statistical modeling of these activities described by their intrinsic physical organic parameters was used to build predictive models for catalyst discovery and to understand their structure–activity relationships. Virtual screening of 102 synthesizable candidates allowed for rapid identification of several ideal catalyst candidates. These statistical models clearly suggest that N-oxyl substructures bearing an adjacent heteroatom are more optimal HAT catalysts compared to the historical focus, phthalimide-N-oxyl, by striking the best balance among all three target experimental properties.
Ni-Catalyzed Enantioselective Desymmetrization: Development of Divergent Acyl and Decarbonylative Cross-Coupling Reactions
JACS
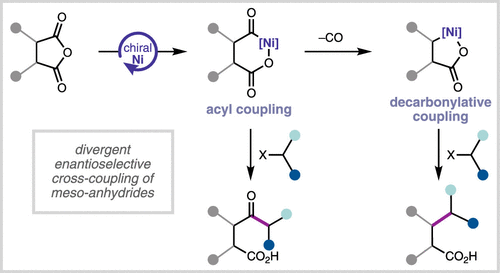
Ángel D. Hernández-Mejías, Alexander M. Shimozono, Avijit Hazra, Sven Richter, Zhengjia Tong, Neil F. Langille, Kyle Quasdorf, Andrew T. Parsons, Matthew S. Sigman, Sarah E. Reisman
Ni-Catalyzed Enantioselective Desymmetrization: Development of Divergent Acyl and Decarbonylative Cross-Coupling Reactions
Ni-catalyzed asymmetric reductive cross-coupling reactions provide rapid and modular access to enantioenriched building blocks from simple electrophile precursors. Reductive coupling reactions that can diverge through a common organometallic intermediate to two distinct families of enantioenriched products are particularly versatile but underdeveloped. Here, we describe the development of a bis(oxazoline) ligand that enables the desymmetrization of meso-anhydrides. When secondary benzylic electrophiles are employed, doubly stereoselective acyl cross-coupling proceeds to give ketone products with catalyst control over three newly formed stereogenic centers. Alternatively, the use of primary alkyl halides in the presence of an additional halogen atom transfer catalyst results in decarbonylative alkylation to give enantioenriched β-alkyl acids. Analysis of reaction rates for a range of both catalysts and substrates supports the notion that tuning the different electrophile activation steps with the two catalysts is required for enhanced reaction performance. These studies illustrate how reaction design can diverge a common Ni-acyl intermediate to either acyl or decarbonylative coupling products and highlight how dual ligand systems can be used to engage unactivated alkyl halides in Ni-catalyzed asymmetric reductive coupling.
Electro-oxidative Deoxyfluorination of Arenes with NEt3·3HF
JOC
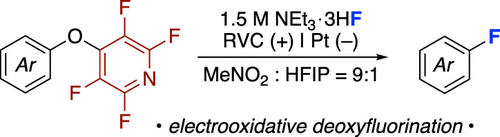
En-Chih Liu, Sabrina M. Reich, Mayank Tanwar, Matthew Neurock, Long Luo, Melanie S. Sanford
Electro-oxidative Deoxyfluorination of Arenes with NEt3·3HF
This report describes the design, development, and optimization of an electrochemical deoxyfluorination of arenes using a tetrafluoropyridine-derived leaving group. NEt3·3HF serves as the fluoride source, and the reactions are conducted using either constant potential or constant current electrolysis in an undivided electrochemical cell. Mechanistic studies support a net oxidative pathway, in which initial single-electron oxidation generates a radical cation intermediate that is trapped by fluoride. The resulting radical undergoes a second oxidation reaction, followed by the loss of the leaving group to yield the fluoroarene product.
Electrifying P(V): Access to Polar and Radical Reactivity
Angewandte Chemie
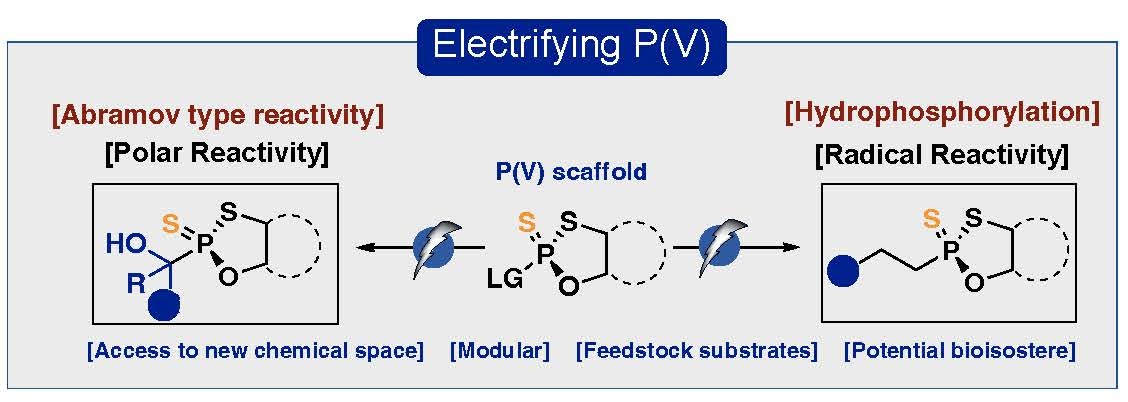
Mahdi Jafarzadeh, Molhm Nassir, Luca Gherardi, Nicholas Raheja, Yu Kawamata, Phil S. Baran
Electrifying P(V): Access to Polar and Radical Reactivity
Electrochemical, fully stereoselective P(V)-radical hydrophosphorylation of olefins and carbonyl compounds using a P(V) reagent is disclosed. By strategically selecting the anode material, radical reactivity is accessible for alkene hydrophosphorylation whereas a polar pathway operates for ketone hydrophosphorylation. The mechanistic intricacies of these chemoselective transformations were explored in-depth.
Stereoselective amino alcohol synthesis via chemoselective electrocatalytic radical cross-couplings
Nature Chemistry
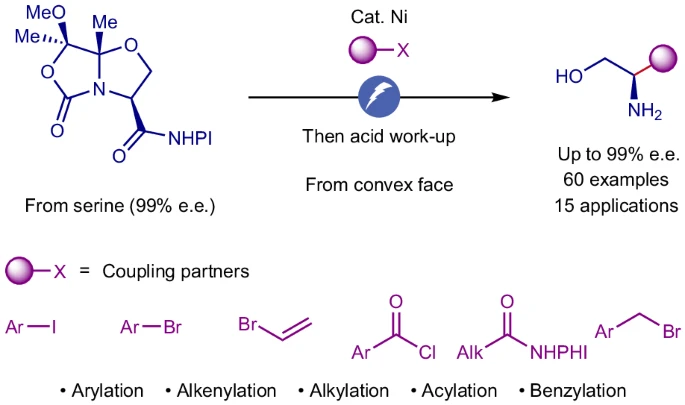
Jiawei Sun, Shuanghu Wang, Kaid C. Harper, Yu Kawamata, Phil S. Baran
Stereoselective amino alcohol synthesis via chemoselective electrocatalytic radical cross-couplings
Amino alcohols are vital in natural products, pharmaceuticals and agrochemicals, and as key building blocks for various applications. Traditional synthesis methods often rely on polar bond retrosynthetic analysis, requiring extensive protecting group manipulations that complicate direct access. Here we show a streamlined approach using a serine-derived chiral carboxylic acid in stereoselective electrocatalytic decarboxylative transformations, enabling efficient access to enantiopure amino alcohols. Unlike conventional strategies, this radical method is both modular and general, offering stereoselective and chemoselective synthesis of diverse substituted amino alcohols. For example, aryl, alkenyl, alkyl and acyl fragments can be coupled efficiently with the serine-derived chiral acid under electrocatalytic decarboxylative conditions. We demonstrate its utility through the rapid synthesis of medicinally important compounds, as well as useful building blocks, highlighting its ability to simplify complex synthetic pathways through entirely different bond disconnections. This electrocatalytic method is robust and scalable, as demonstrated in a 72-gram-scale flow reaction.
Utility of Immobilized Metal Salens as Electrocatalysts: Fuel Cells and Organic Electrosynthesis
ChemElectroChem
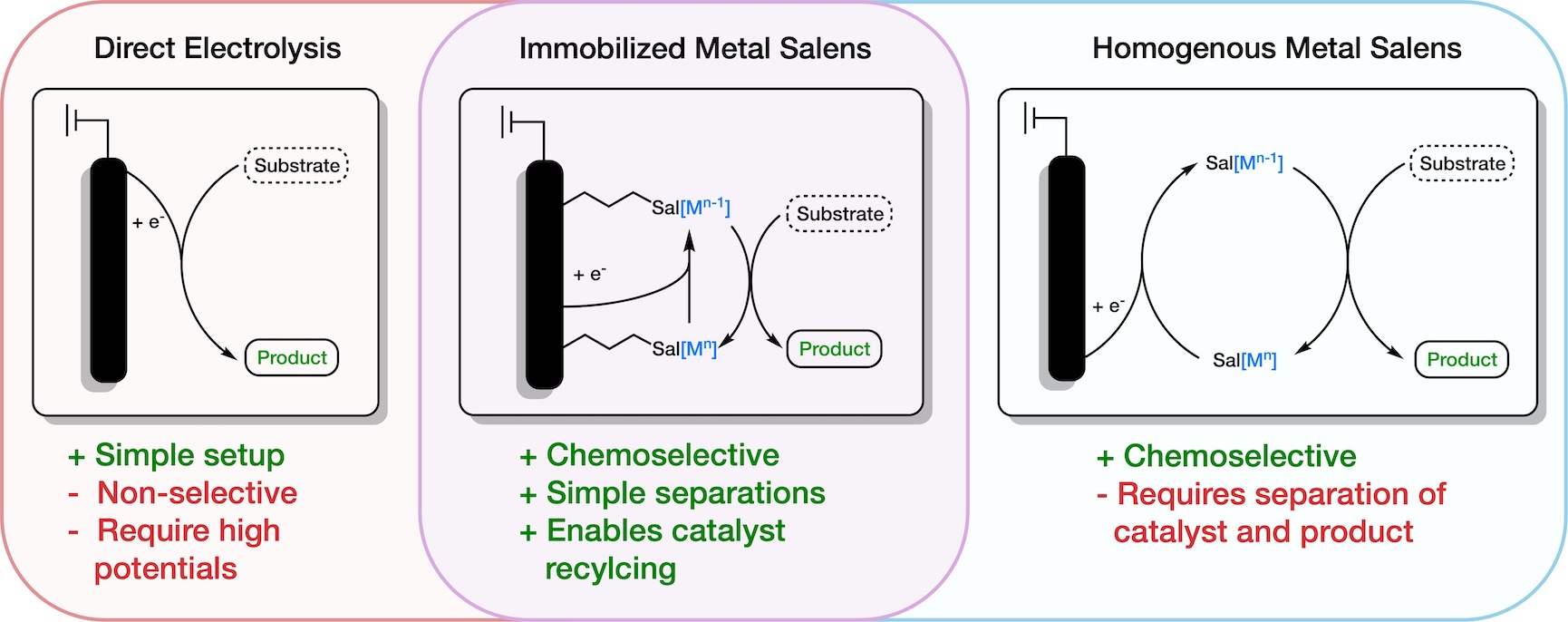 Zachary A. Nguyen, Shelley D. Minteer
Zachary A. Nguyen, Shelley D. Minteer
Utility of Immobilized Metal Salens as Electrocatalysts: Fuel Cells and Organic Electrosynthesis
There have been significant advancements in the electrosynthesis of fuels and organic molecules, making it an increasingly sustainable and cost-effective alternative to traditional chemical redox reagents. Early versions of these systems faced challenges in chemoselectivity due to high applied overpotentials, which have been mitigated with the introduction of molecular electrocatalysts, like metal salens (MSalens). These MSalens reduce the required overpotentials, increase turnover numbers (TON), and have simple modularity within their ligand structure allowing for tunable selectivity. While these MSalen electrocatalysts are typically used homogeneously for engineering simplicity, downstream separations are often costly and time-consuming. Immobilization of MSalens addresses these issues by enabling synthesis at lower potentials, achieving high selectivity, and facilitating straightforward separations. This review explores the application of MSalens in electrosynthesis and immobilized molecular electrocatalysts in organic electrosynthesis.
Pyrolytic Carbon: An Inexpensive, Robust, and Versatile Electrode for Synthetic Organic Electrochemistry
Angewandte Chemie
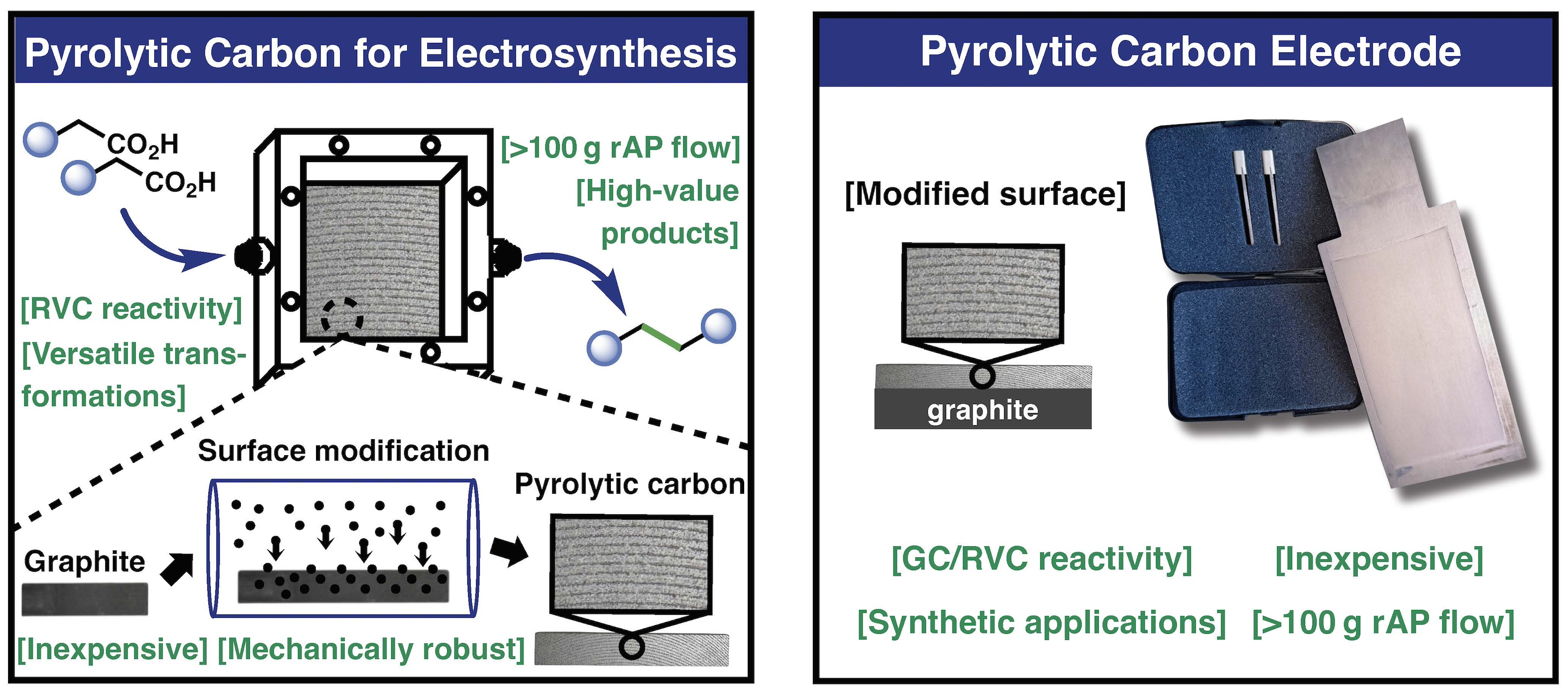
Tamara El-Hayek Ewing, Nils Kurig, Yoshio Robert Yamaki, Jiawei Sun, Timothy R Knowles, Asha Gollapudi, Yu Kawamata, Phil S. Baran
Pyrolytic Carbon: An Inexpensive, Robust, and Versatile Electrode for Synthetic Organic Electrochemistry
Synthetic organic electrochemistry is recognized as one of the most sustainable forms of redox chemistry that can enable a wide variety of useful transformations. In this study, readily prepared pyrolytic carbon electrodes are explored in several powerful rAP transformations as well as C–C and C–N bond forming reactions. Pyrolytic carbon provides an alternative to classic amorphous carbon-based materials that are either expensive or ill-suited to large-scale flow reactions.
The Electrochemical Peroxydisulfate-Oxalate Autocatalytic Reaction
JACS
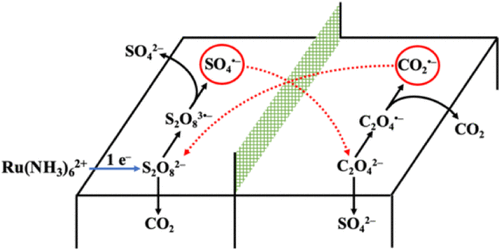
Jordyn N. Janusz, Joshua A. Beeler, Seyyedamirhossein Hosseini, Mayank Tanwar, Rui Zeng, Hongsen Wang, Héctor D. Abruña, Matthew Neurock, Henry S. White
The Electrochemical Peroxydisulfate-Oxalate Autocatalytic Reaction
Aqueous solutions containing both the strong oxidant, peroxydisulfate (S2O82–), and the strong reductant, oxalate (C2O42–), are thermodynamically unstable due to the highly exothermic homogeneous redox reaction: S2O82– + C2O42– → 2 SO42– + 2 CO2 (ΔG0 = −490 kJ/mol). However, at room temperature, this reaction does not occur to a significant extent over the time scale of a day due to its inherently slow kinetics. We demonstrate that the S2O82–/C2O42– redox reaction occurs rapidly, once initiated by the Ru(NH3)62+-mediated 1e– reduction of S2O82– to form S2O83•–, which rapidly undergoes bond cleavage to form SO42– and the highly oxidizing radical SO4•–. Theoretically, the mediated electrochemical generation of a single molecule of S2O83•– can initiate an autocatalytic cycle that consumes both S2O82– and C2O42– in bulk solution. Several experimental demonstrations of S2O82–/C2O42– autocatalysis are presented. Differential electrochemical mass spectrometry measurements demonstrate that CO2 is generated in solution for at least 10 min following a 30-s initiation step. Quantitative bulk electrolysis of S2O82– in solutions containing excess C2O42– is initiated by electrogeneration of immeasurably small quantities of S2O83•–. Capture of CO2 as BaCO3 during electrolysis additionally confirms the autocatalytic generation of CO2. First-principles density functional theory calculations, ab initio molecular dynamics simulations, and finite difference simulations of cyclic voltammetric responses are presented that support and provide additional insights into the initiation and mechanism of S2O82–/C2O42– autocatalysis. Preliminary evidence indicates that autocatalysis also results in a chemical traveling reaction front that propagates into the solution normal to the planar electrode surface.
Developing Microelectrode Arrays for the Point-of-Care Multiplex Detection of Metabolites
Analytical Chemistry
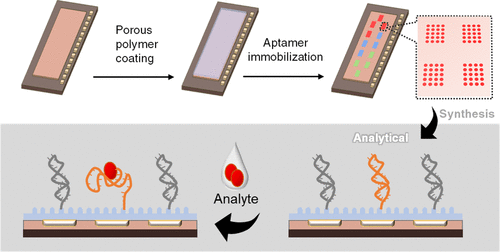
Yu-Chia Chang, Benoit Arnould, Jennifer M. Heemstra, Kevin D. Moeller
Developing Microelectrode Arrays for the Point-of-Care Multiplex Detection of Metabolites
DNA-aptamer-functionalized electrode arrays can provide an intriguing method for detecting pathogen-derived exometabolites. This work addresses the limitations of previous aptamer-based pathogen detection methods by introducing a novel surface design that bridges the gap between initial efforts in this area and the demands of a point-of-care device. Specifically, the use of a diblock copolymer coating on a high-density microelectrode array and Cu-mediated cross coupling reactions that allow for the exclusive functionalization of that coating by any electrode or set of electrodes in the array provides a device that is stable for 1 year and compatible with the multiplex detection of small-molecule targets. The new chemistry developed allows one to take advantage of a large number of electrodes in the array with one experiment described herein capitalizing on the use of 960 individually addressable electrodes.
Reductive samarium (electro)catalysis enabled by SmIII-alkoxide protonolysis
Science
Emily A. Boyd, Chungkeun Shin, David J. Charboneau, Jonas C. Peters, and Sarah E. Reisman
Reductive samarium (electro)catalysis enabled by SmIII-alkoxide protonolysis
Samarium diiodide (SmI2) is a privileged, single-electron reductant deployed in diverse synthetic settings. However, generalizable methods for catalytic turnover remain elusive because of the well-known challenge associated with cleaving strong SmIII–O bonds. Prior efforts have focused on the use of highly reactive oxophiles to enable catalyst turnover. However, such approaches give rise to complex catalyst speciation and intrinsically limit the synthetic scope. Herein, we leveraged a mild and selective protonolysis strategy to achieve samarium-catalyzed, intermolecular reductive cross-coupling of ketones and acrylates with broad scope. The modularity of our approach allows rational control of selectivity based on solvent, pKa (where Ka is the acid dissociation constant), and the samarium coordination sphere and provides a basis for future developments in catalytic and electrocatalytic lanthanide chemistry.
CdS Quantum Dot Gels as a Direct Hydrogen Atom Transfer Photocatalyst for C-H Activation
Angewandte Chemie
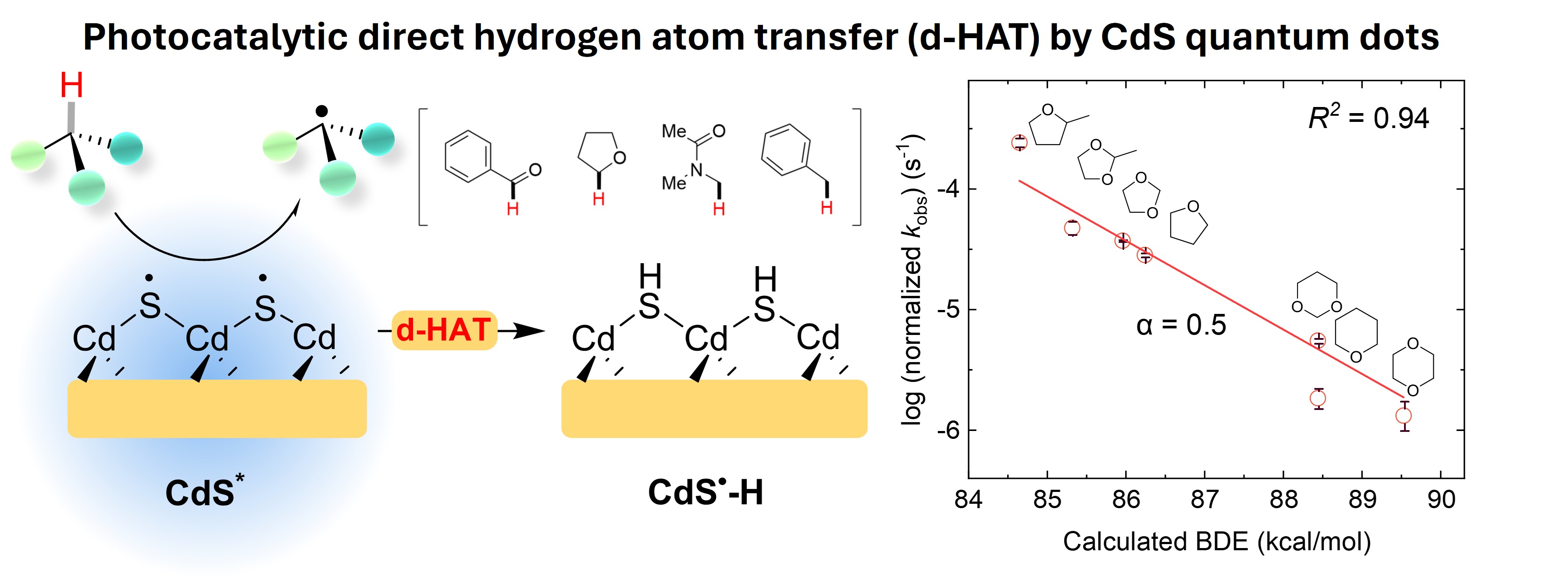
CdS Quantum Dot Gels as a Direct Hydrogen Atom Transfer Photocatalyst for C-H Activation
Here, we report CdS quantum dot (QD) gels, a three-dimensional network of interconnected CdS QDs, as a new type of direct hydrogen atom transfer (d-HAT) photocatalyst for C-H activation. We discovered that the photoexcited CdS QD gel could generate various neutral radicals, including α-amido, heterocyclic, acyl, and benzylic radicals, from their corresponding stable molecular substrates, including amides, thio/ethers, aldehydes, and benzylic compounds. Its C-H activation ability imparts a broad substrate and reaction scope. The mechanistic study reveals that this reactivity is intrinsic to CdS materials, and the neutral radical generation did not proceed via the conventional sequential electron transfer and proton transfer pathway. Instead, the C-H bonds are activated by the photoexcited CdS QD gel via a d-HAT mechanism. This d-HAT mechanism is supported by the linear correlation between the logarithm of the C-H bond activation rate constant and the C-H bond dissociation energy (BDE) with a Brønsted slope α = 0.5. Our findings expand the currently limited direct hydrogen atom transfer photocatalysis toolbox and provide new possibilities for photocatalytic C-H activation.
Improved Electrosynthesis of Biomass Derived Furanic Compounds via Nitroxyl Radical Redox Mediation
Chem & Bio Engineering
Emily Carroll, Sarah L. Parker, Anna Fukushima, Sophie Downey, Delaney Miller, Zachary A. Nguyen, Dylan G. Boucher, and Shelley D. Minteer
Improved Electrosynthesis of Biomass Derived Furanic Compounds via Nitroxyl Radical Redox Mediation
Biomass is an abundantly available, underutilized feedstock for the production of bulk and fine chemicals, polymers, and sustainable and biodegradable plastics that are traditionally sourced from petrochemicals. Among potential feedstocks, 2,5-furan dicarboxylic acid (FDCA) stands out for its potential to be converted to higher-value polymeric materials such as polyethylene furandicarboxylate (PEF), a bio-based plastic alternative. In this study, the sustainable, electrocatalytic oxidation of stable furan molecule 2,5-bis(hydroxymethyl)furan (BHMF) to FDCA is investigated using a variety of TEMPO derivative electrocatalysts in a mediated electrosynthetic reaction. Three TEMPO catalysts (acetamido-TEMPO, methoxy-TEMPO, and TEMPO) facilitate full conversion to FDCA in basic conditions with >90% yield and >100% Faradaic efficiency. The remaining three TEMPO catalysts (hydroxy-TEMPO, oxo-TEMPO, and amino-TEMPO) all perform intermediate oxidation of BHMF in basic conditions but do not facilitate full conversion to FDCA. On the basis of pH studies completed on all TEMPO derivatives to assess their electrochemical reversibility and response to substrate, pH and reversibility play significant roles in the catalytic ability of each catalyst, which directly influences catalyst turnover and product formation. More broadly, this study also highlights the importance of an effective and rapid electroanalytical workflow in mediated electrosynthetic reactions, demonstrating how voltammetric catalyst screening can serve as a useful tool for predicting the reactivity and efficacy of a catalyst–substrate electrochemical system.
Investigation of the Electrocatalytic Reduction of Peroxydisulfate Using Scanning Electrochemical Microscopy
Analytical Chemistry
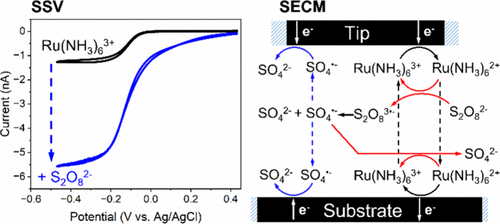
Seyyedamirhossein Hosseini, Gergely T. Solymosi, and Henry S. White
Investigation of the Electrocatalytic Reduction of Peroxydisulfate Using Scanning Electrochemical Microscopy
The elementary steps of the electrocatalytic reduction of S2O82– using the Ru(NH3)63+/2+
redox couple were investigated using scanning electrochemical microscopy (SECM) and
steady-state voltammetry (SSV). SECM investigations were carried out in a 0.1 M KCl
solution using a 3.5 μm radius carbon ultramicroelectrode (UME) as the SECM tip and
a 25 μm radius platinum UME as the substrate electrode. Approach curves were recorded
in the positive feedback mode of SECM by reducing Ru(NH3)63+ at the tip electrode
and oxidizing Ru(NH3)62+ at the substrate electrode, as a function of the tip–substrate
separation and S2O82– concentration. The one-electron reaction between electrogenerated
Ru(NH3)62+ and S2O82– yields the unstable S2O83•–, which rapidly dissociates to produce
highly oxidizing SO4•–. Because SO4•– is such a strongly oxidizing species, it can
be further reduced at both the tip and the substrate, or it can react with Ru(NH3)62+
to regenerate Ru(NH3)63+. SECM approach curves display a complex dependence on the
tip–substrate distance, d, due to redox mediation reactions at both the tip and the
substrate. Finite element method (FEM) simulations of both SECM approach curves and
SSV confirm a previously proposed mechanism for the mediated reduction of S2O82– using
the Ru(NH3)63+/2+ redox couple. Our results provide a lower limit for dissociation
rate constant of S2O83•– (∼1 × 106 s–1), as well as the rate constants for electron
transfer between SO4•– and Ru(NH3)62+ (∼1 × 109 M–1 s–1) and between S2O82– and Ru(NH3)62+
(∼7 × 105 M–1 s–1).
Autonomous closed-loop mechanistic investigation of molecular electrochemistry via automation
Nature Communications

Hongyuan Sheng, Jingwen Sun, Oliver Rodríguez, Benjamin B. Hoar, Weitong Zhang, Danlei Xiang, Tianhua Tang, Avijit Hazra, Daniel S. Min, Abigail G. Doyle, Matthew S. Sigman, Cyrille Costentin, Quanquan Gu, Joaquín Rodríguez-López & Chong Liu
Autonomous closed-loop mechanistic investigation of molecular electrochemistry via automation
Electrochemical research often requires stringent combinations of experimental parameters that are demanding to manually locate. Recent advances in automated instrumentation and machine-learning algorithms unlock the possibility for accelerated studies of electrochemical fundamentals via high-throughput, online decision-making. Here we report an autonomous electrochemical platform that implements an adaptive, closed-loop workflow for mechanistic investigation of molecular electrochemistry. As a proof-of-concept, this platform autonomously identifies and investigates an EC mechanism, an interfacial electron transfer (E step) followed by a solution reaction (C step), for cobalt tetraphenylporphyrin exposed to a library of organohalide electrophiles. The generally applicable workflow accurately discerns the EC mechanism’s presence amid negative controls and outliers, adaptively designs desired experimental conditions, and quantitatively extracts kinetic information of the C step spanning over 7 orders of magnitude, from which mechanistic insights into oxidative addition pathways are gained. This work opens opportunities for autonomous mechanistic discoveries in self-driving electrochemistry laboratories without manual intervention.
Benchmarking Trisaminocyclopropeniums as Mediators for Anodic Oxidation Reactions
JOC

Sabrina N. Carneiro, Joshua D. Laffoon, Long Luo, and Melanie S. Sanford
Benchmarking Trisaminocyclopropeniums as Mediators for Anodic Oxidation Reactions
This report benchmarks a tris(amino)cyclopropenium (TAC) salt as an electron-transfer mediator for anodic oxidation reactions in comparison to two known mediators: a triarylamine and a triarylimidazole derivative. The three mediators have redox potentials, diffusion coefficients, and heterogeneous electron transfer rates similar to those of glassy carbon electrodes in acetonitrile/KPF6. However, they differ significantly in their performance in two electro-organic reactions: anodic fluorination of a dithiane and anodic oxidation of 4-methoxybenzyl alcohol. These differences are rationalized based on variable stability in the presence of reaction components (e.g., NEt3·3HF, lutidine, and Cs2CO3) as well as very different rates of electron transfer with the organic substrate. Overall, this work highlights the advantages and disadvantages of each mediator and provides a foundation for expanding the applications of TACs in electro-organic synthesis moving forward.
Connecting Interfacial Mechanical Adhesion, Efficiency, and Operational Stability in High Performance Inverted Perovskite Solar Cells
ACS Energy Letters

Zhenghong Dai, Shuai You, Dwaipayan Chakraborty, Shunran Li, Yadong Zhang, Anush Ranka, Stephen Barlow, Joseph J. Berry, Seth R. Marder, Peijun Guo, Yue Qi, Kai Zhu, and Nitin P. Padture
Connecting Interfacial Mechanical Adhesion, Efficiency, and Operational Stability in High Performance Inverted Perovskite Solar Cells
Carbazole-based self-assembled monolayers (SAMs) at the interface between the metal-halide perovskite (MHP) and the transparent conducting oxide (TCO) serve the function of hole-transport layers in p-i-n “inverted” perovskite solar cells (PSCs). Here we show that the use of an iodine-terminated carbazole-based SAM increases the interfacial mechanical adhesion dramatically (2.6-fold) and that this is responsible for substantial improvements in the interfacial morphology, photocarrier transport, and operational stability. While the improved morphology and optoelectronic properties impart high efficiency (up to 25.39%) to the PSCs, the enhanced adhesion suppresses nucleation and propagation of pores/cracks during PSC operation, resulting in the retention of 96% of the initial efficiency after 1000 h of continuous-illumination testing at the maximum power-point. This demonstrates the strong connection between judicious interfacial adhesion toughening and simultaneous enhancement in the efficiency and operational stability of p-i-n PSCs, with broader implications for the reliability and durability of perovskite photovoltaics before they can be commercialized.
Bioelectrocatalysis for synthetic applications: Utilities and challenges
Current Opinion in Electrochemistry
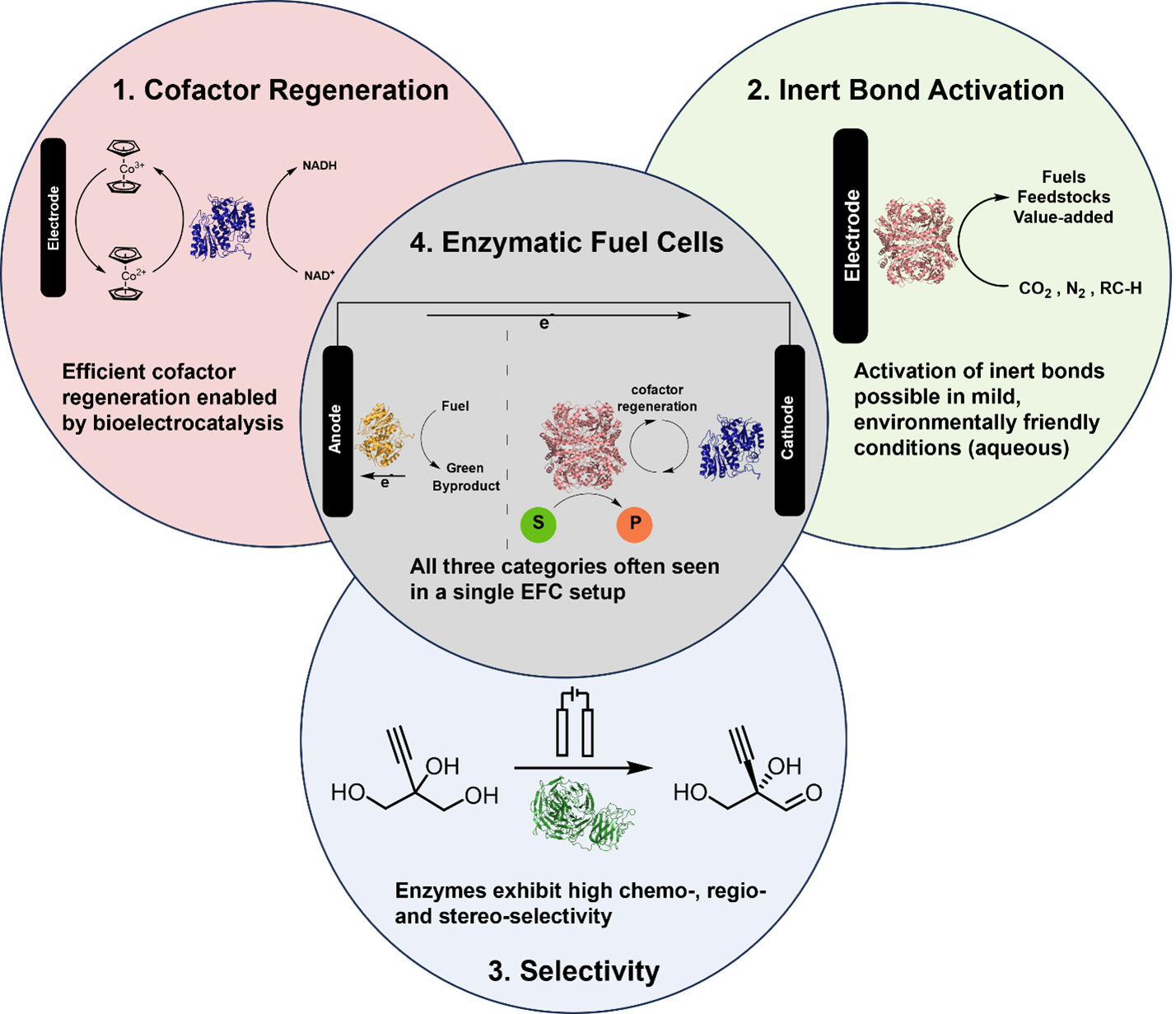 Kevin M. McFadden, Luke G. Kays, Dylan G. Boucher, and Shelley D. Minteer
Kevin M. McFadden, Luke G. Kays, Dylan G. Boucher, and Shelley D. Minteer
Bioelectrocatalysis for synthetic applications: Utilities and challenges
The pairing of enzymes with electrochemistry to accomplish bioelectrocatalysis enables the inherent advantages of each to be harnessed toward efficient green synthesis of fuels, feedstocks, agrochemicals and pharmaceuticals. This article seeks to describe the current advantages of bioelectrocatalysis for synthesis applications and recent research to further expand the scope of bioelectrocatalysis. We focus on recent progress in bioelectrocatalytic cofactor regeneration, selectivity, inert bond activation under aqueous conditions and how all three can be applied in enzymatic fuel cells. We also cover the current strategies to overcome the most prominent challenges in the field, with an emphasis on enzyme stability, cascades for increasing product complexity and directed evolution.
Simplified Modular Access to Enantiopure 1,2-Aminoalcohols via Ni-Electrocatalytic Decarboxylative Arylation
JACS
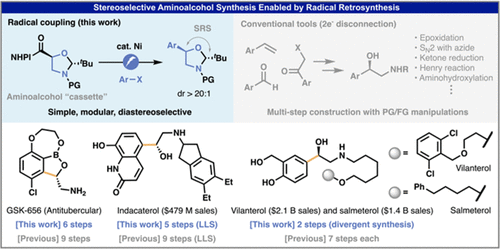
Jiawei Sun, Hirofumi Endo, Megan A. Emmanuel, Martins S. Oderinde, Yu Kawamata, and Phil S. Baran
Simplified Modular Access to Enantiopure 1,2-Aminoalcohols via Ni-Electrocatalytic Decarboxylative Arylation
Chiral aminoalcohols are omnipresent in bioactive compounds. Conventional strategies to access this motif involve multiple-step reactions to install the requisite functionalities stereoselectively using conventional polar bond analysis. This study reveals that a simple chiral oxazolidine-based carboxylic acid can be readily transformed to substituted chiral aminoalcohols with high stereochemical control by Ni-electrocatalytic decarboxylative arylation. This general, robust, and scalable coupling can be used to synthesize a variety of medicinally important compounds, avoiding protecting and functional group manipulations, thereby dramatically simplifying their preparation.
Simplifying Access to Targeted Protein Degraders via Nickel Electrocatalytic Cross-Coupling
Angewandte Chemie
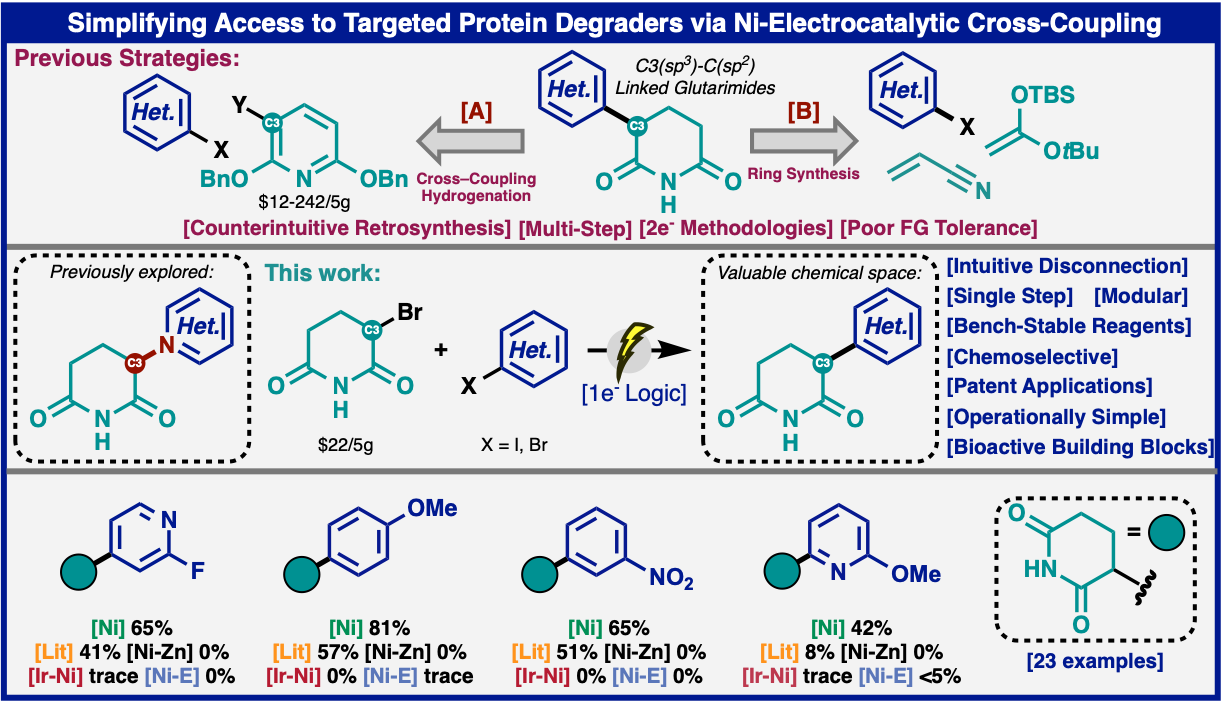
Philipp Neigenfind, Luca Massaro, Áron Péter, Andrew P. Degnan, Megan A. Emmanuel, Martins S. Oderinde, Chi He, David Peters, Tamara El-Hayek Ewing, Yu Kawamata, Phil S. Baran
Simplifying Access to Targeted Protein Degraders via Nickel Electrocatalytic Cross-Coupling
C−C linked glutarimide-containing structures with direct utility in the preparation of cereblon-based degraders (PROTACs, CELMoDs) can be assessed in a single step from inexpensive, commercial α-bromoglutarimide through a unique Brønsted-acid assisted Ni-electrocatalytic approach. The reaction tolerates a broad array of functional groups that are historically problematic and can be applied to the simplified synthesis of dozens of known compounds that have only been procured through laborious, wasteful, multi-step sequences. The reaction is scalable in both batch and flow and features a trivial procedure wherein the most time-consuming aspect of reaction setup is weighing out the starting materials.
Nickel-Electrocatalytic Decarboxylative Arylation to Access Quaternary Centers
Angewandte Chemie
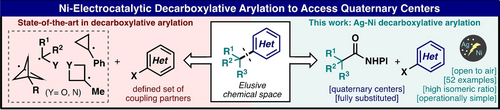
Gabriele Laudadio, Philipp Neigenfind, Áron Péter, Camille Z. Rubel, Megan A. Emmanuel, Martins S. Oderinde, Tamara El-Hayek Ewing, Maximilian D. Palkowitz, Jack L. Sloane, Kevin W. Gillman, Daniel Ridge, Michael D. Mandler, Philippe N. Bolduc, Michael C. Nicastri, Benxiang Zhang, Sebastian Clementson, Nadia Nasser Petersen, Pablo Martín-Gago, Pavel Mykhailiuk, Keary M. Engle, Phil S. Baran
Nickel-Electrocatalytic Decarboxylative Arylation to Access Quaternary Centers
There is a pressing need, particularly in the field of drug discovery, for general methods that will enable direct coupling of tertiary alkyl fragments to (hetero)aryl halides. Herein a uniquely powerful and simple set of conditions for achieving this transformation with unparalleled generality and chemoselectivity is disclosed. This new protocol is placed in context with other recently reported methods, applied to simplify the routes of known bioactive building blocks molecules, and scaled up in both batch and flow. The role of pyridine additive as well as the mechanism of this reaction are interrogated through Cyclic Voltammetry studies, titration experiments, control reactions with Ni(0) and Ni(II)-complexes, and ligand optimization data. Those studies indicate that the formation of a BINAPNi(0) is minimized and the formation of an active pyridine-stabilized Ni(I) species is sustained during the reaction. Our preliminary mechanistic studies ruled out the involvement of Ni(0) species in this electrochemical cross-coupling, which is mediated by Ni(I) species via a Ni(I)-Ni(II)-Ni(III)-Ni(I) catalytic cycle.
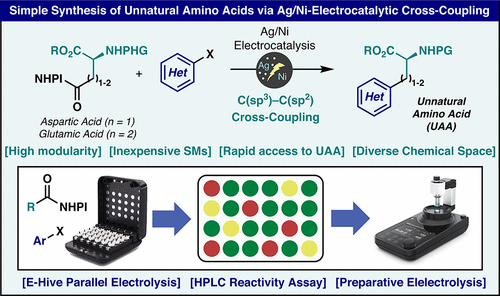
Synthesis of Unnatural Amino Acids via Ni/Ag Electrocatalytic Cross-Coupling
Organic Letters
 Gabriele Laudadio, Philipp Neigenfind, Rajesh Chebolu, Vanna D. Blasczak, Shambabu
Joseph Maddirala, Maximilian D. Palkowitz, Philippe N. Bolduc, Michael C. Nicastri,
Ravi Kumar Puthukanoori, Bheema Rao Paraselli, and Phil S. Baran
Gabriele Laudadio, Philipp Neigenfind, Rajesh Chebolu, Vanna D. Blasczak, Shambabu
Joseph Maddirala, Maximilian D. Palkowitz, Philippe N. Bolduc, Michael C. Nicastri,
Ravi Kumar Puthukanoori, Bheema Rao Paraselli, and Phil S. Baran
Synthesis of Unnatural Amino Acids via Ni/Ag Electrocatalytic Cross-Coupling
A simple protocol is outlined herein for rapid access to enantiopure unnatural amino acids (UAAs) from trivial glutamate and aspartate precursors. The method relies on Ag/Ni-electrocatalytic decarboxylative coupling and can be rapidly conducted in parallel (24 reactions at a time) to ascertain coupling viability followed by scale-up for the generation of useful quantities of UAAs for exploratory studies.
A Guide to Troubleshooting Metal Sacrificial Anodes for Organic Electrosynthesis
Chemical Science
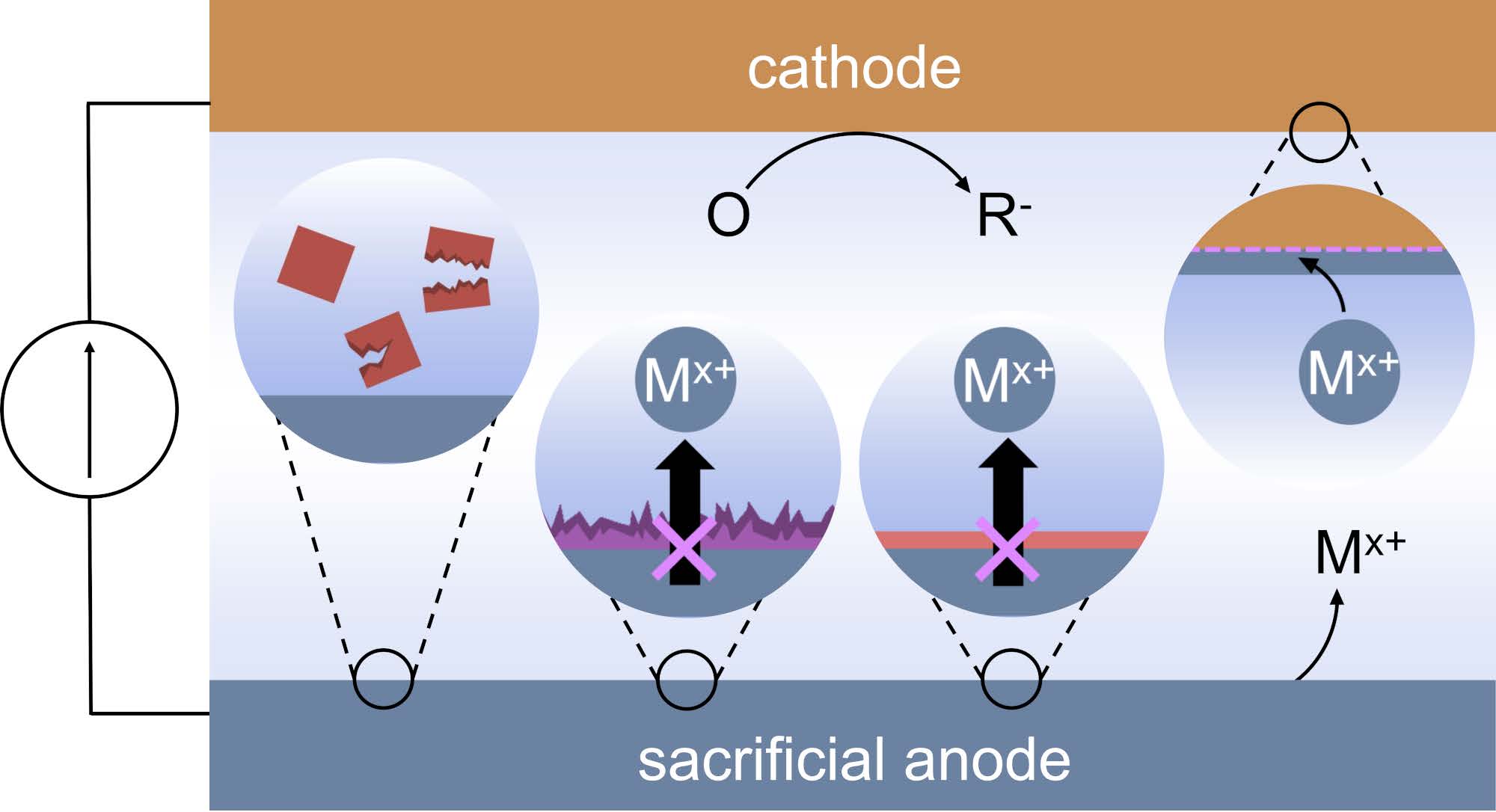
Skyler D. Ware, Wendy Zhang, Weiyang Guan, Song Lin and Kimberly A. See
A Guide to Troubleshooting Metal Sacrificial Anodes for Organic Electrosynthesis
Sacrificial anodes enable reductive electrosynthesis by charge-balancing the reaction of interest occurring at the cathode via oxidation of the bulk metal anode. While this metal oxidation is often assumed to be straightforward and innocent relative to the chemistry at the cathode, in reality non-ideal processes at the anode can interfere with the reductive chemistry. In this perspective, we highlight several common challenges that arise when using sacrificial anodes, and we propose experiments to diagnose and troubleshoot each issue. We anticipate that a thorough understanding of sacrificial anode chemistry will streamline reaction optimization and expand the chemical space available for organic electrosynthesis.
Electrocatalytic Asymmetric Nozaki–Hiyama–Kishi Decarboxylative Coupling: Scope, Applications, and Mechanism
JACS
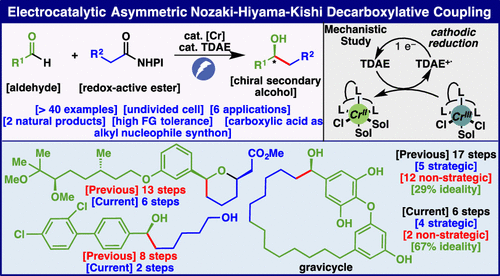 Yang Gao, Baiyang Jiang, Nathan C. Friede, Arianne C. Hunter, Dylan G. Boucher, Shelley D. Minteer, Matthew S. Sigman, Sarah E. Reisman, and Phil S. Baran
Yang Gao, Baiyang Jiang, Nathan C. Friede, Arianne C. Hunter, Dylan G. Boucher, Shelley D. Minteer, Matthew S. Sigman, Sarah E. Reisman, and Phil S. Baran
Electrocatalytic Asymmetric Nozaki–Hiyama–Kishi Decarboxylative Coupling: Scope, Applications, and Mechanism
The first general enantioselective alkyl-Nozaki–Hiyama–Kishi (NHK) coupling reactions are disclosed herein by employing a Cr-electrocatalytic decarboxylative approach. Using easily accessible aliphatic carboxylic acids (via redox-active esters) as alkyl nucleophile synthons, in combination with aldehydes and enabling additives, chiral secondary alcohols are produced in a good yield with high enantioselectivity under mild reductive electrolysis. This reaction, which cannot be mimicked using stoichiometric metal or organic reductants, tolerates a broad range of functional groups and is successfully applied to dramatically simplify the synthesis of multiple medicinally relevant structures and natural products. Mechanistic studies revealed that this asymmetric alkyl e-NHK reaction was enabled by using catalytic tetrakis(dimethylamino)ethylene, which acts as a key reductive mediator to mediate the electroreduction of the CrIII/chiral ligand complex.
Anodic Cyclizations, Densely Functionalized Synthetic Building Blocks, and the Importance of Recent Mechanistic Observations
JOC
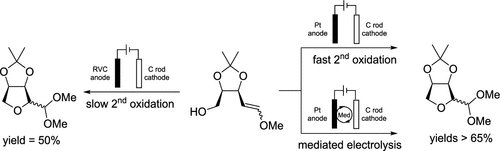
Ruby Krueger, Enqi Feng, Polina Barzova, Noah Lieberman, Song Lin, and Kevin D. Moeller
Anodic Cyclizations, Densely Functionalized Synthetic Building Blocks, and the Importance of Recent Mechanistic Observations
Anodic cyclization reactions can provide a versatile method for converting newly obtained chiral lactols to densely functionalized cyclic building blocks. The method works by first converting the lactol into an electron-rich olefin and then oxidatively generating a radical cation that is trapped by a nucleophile. Historically, such reactions have benefited from the use of less polar radical cations when the trapping nucleophile is a heteroatom and more polar radical cations when the reaction forms C–C bonds. This forced one to optimize underperforming reactions by resynthesizing the substrate. Here, we show that by taking advantage of methods that serve to drive a reversible initial cyclization reaction toward the product, this dichotomy and need to manipulate the substrate can be avoided. Two such methods were utilized: a faster second oxidation step and a mediated electrolysis. Both led to successful cyclizations using a polar radical cation and heteroatom nucleophiles.
Electroreductive Synthesis of Nickel(0) Complexes
Angewandte Chemie
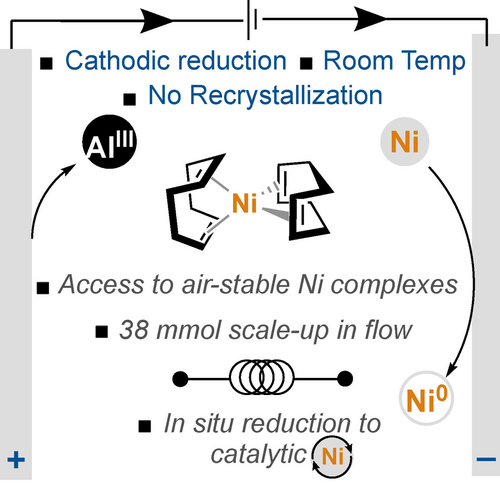
Camille Z. Rubel, Yilin Cao, Tamara El-Hayek Ewing, Gabriele Laudadio, Gregory L. Beutner, Steven R. Wisniewski, Xiangyu Wu, Phil S. Baran, Julien C. Vantourout, Keary M. Engle
Electroreductive Synthesis of Nickel(0) Complexes
Over the last fifty years, the use of nickel catalysts for facilitating organic transformations has skyrocketed. Nickel(0) sources act as useful precatalysts because they can enter a catalytic cycle through ligand exchange, without needing to undergo additional elementary steps. However, most Ni(0) precatalysts are synthesized with stoichiometric aluminum–hydride reductants, pyrophoric reagents that are not atom-economical and must be used at cryogenic temperatures. Here, we demonstrate that Ni(II) salts can be reduced on preparative scale using electrolysis to yield a variety of Ni(0) and Ni(II) complexes that are widely used as precatalysts in organic synthesis, including bis(1,5-cyclooctadiene)nickel(0) [Ni(COD)2]. This method overcomes the reproducibility issues of previously reported methods by standardizing the procedure, such that it can be performed anywhere in a robust manner. It can be transitioned to large scale through an electrochemical recirculating flow process and extended to an in situ reduction protocol to generate catalytic amounts of Ni(0) for organic transformations. We anticipate that this work will accelerate adoption of preparative electrochemistry for the synthesis of low-valent organometallic complexes in academia and industry.
Advanced Electroanalysis for Electrosynthesis
ACS Organic & Inorganic Au
 Monica Brachi, Wassim El Housseini, Kevin Beaver, Rohit Jadhav, Ashwini Dantanarayana,
Dylan G. Boucher, and Shelley D. Minteer
Monica Brachi, Wassim El Housseini, Kevin Beaver, Rohit Jadhav, Ashwini Dantanarayana,
Dylan G. Boucher, and Shelley D. Minteer
Advanced Electroanalysis for Electrosynthesis
Electrosynthesis is a popular, environmentally friendly substitute for conventional organic methods. It involves using charge transfer to stimulate chemical reactions through the application of a potential or current between two electrodes. In addition to electrode materials and the type of reactor employed, the strategies for controlling potential and current have an impact on the yields, product distribution, and reaction mechanism. In this Review, recent advances related to electroanalysis applied in electrosynthesis were discussed. The first part of this study acts as a guide that emphasizes the foundations of electrosynthesis. These essentials include instrumentation, electrode selection, cell design, and electrosynthesis methodologies. Then, advances in electroanalytical techniques applied in organic, enzymatic, and microbial electrosynthesis are illustrated with specific cases studied in recent literature. To conclude, a discussion of future possibilities that intend to advance the academic and industrial areas is presented.
Enabling Al sacrificial anodes in tetrahydrofuran electrolytes for reductive electrosynthesis
Chemical Science
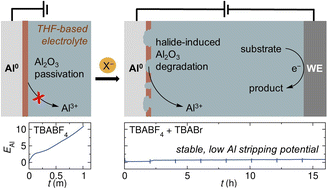
Wendy Zhang, Weiyang Guan, Yi Wang, Song Lin and Kimberly A. See
Enabling Al sacrificial anodes in tetrahydrofuran electrolytes for reductive electrosynthesis
Al is widely used as a sacrificial anode in organic electrosynthesis. However, there remains a notable knowledge gap in the understanding of Al anode interface chemistry under electrolysis conditions. We hypothesize that Al interfacial chemistry plays a pivotal role in the discernible bias observed in solvent selections for reductive electrosynthesis. The majority of existing methodologies that employ an Al sacrificial anode use N,N-dimethylformamide (DMF) as the preferred solvent, with only isolated examples of ethereal solvents such as tetrahydrofuran (THF). Given the crucial role of the solvent in determining the efficiency and selectivity of an organic reaction, limitations on solvent choice could significantly hinder substrate reactivity and impede the desired transformations. In this study, we aim to understand the Al metal interfaces and manipulate them to improve the performance of an Al sacrificial anode in THF-based electrolytes. We have discovered that the presence of halide ions (Cl−, Br−, I−) in the electrolyte is crucial for efficient Al stripping. By incorporating halide additive, we achieve bulk Al stripping in THF-based electrolytes and successfully improve the cell potentials of electrochemically driven reductive methodologies. This study will encourage the use of ethereal solvents in systems using Al sacrificial anodes and guide future endeavors in optimizing electrolytes for reductive electrosynthesis.
Tailoring Ag Electron Donating Ability for Organohalide Reduction: A Bilayer Electrode Design
Langmuir
 Ali Abbaspourtamijani, Dwaipayan Chakraborty, Henry Sheldon White, Matthew Neurock, and Yue Qi
Ali Abbaspourtamijani, Dwaipayan Chakraborty, Henry Sheldon White, Matthew Neurock, and Yue Qi
Tailoring Ag Electron Donating Ability for Organohalide Reduction: A Bilayer Electrode Design
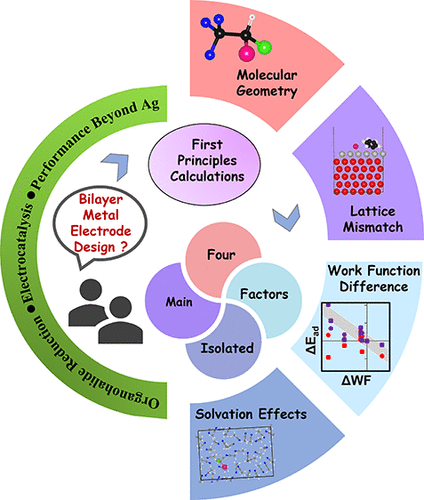
Electrochemical reduction of organohalides provides a green approach in the reduction of environmental pollutants, the synthesis of new organic molecules, and many other applications. The presence of a catalytic electrode can make the process more energetically efficient. Ag is known to be a very good electrode for the reduction of a wide range of organohalides. Herein, we examine the elementary adsorption and reaction steps that occur on Ag and the changes that result from changes in the Ag-coated metal, strain in Ag, solvent, and substrate geometry. The results are used to develop an electrode design strategy that can possibly be used to further increase the catalytic activity of pure Ag electrodes. We have shown how epitaxially depositing one to three layers of Ag on catalytically inert or less active support metal can increase the surface electron donating ability, thus increasing the adsorption of organic halide and the catalytic activity. Many factors, such as molecular geometry, lattice mismatches, work function, and solvents, contribute to the adsorption of organic halide molecules over the bilayer electrode surface. To isolate and rank these factors, we examined three model organic halides, namely, halothane, bromobenzene (BrBz), and benzyl bromide (BzBr) adsorption on Ag/metal (metal = Au, Bi, Pt, and Ti) bilayer electrodes in both vacuum and acetonitrile (ACN) solvent. The different metal supports offer a range of lattice mismatches and work function differences with Ag. Our calculations show that the surface of Ag becomes more electron donating and accessible to adsorption when it forms a bilayer with Ti as it has a lower work function and almost zero lattice mismatch with Ag. We believe this study will help to increase the electron donating ability of the Ag surface by choosing the right metal support, which in turn can improve the catalytic activity of the working electrode.
Three-Component Cross-Electrophile Coupling: Regioselective Electrochemical Dialkylation of Alkenes
JACS
Lingxiang Lu, Yi Wang, Wendy Zhang, Wen Zhang, Kimberly A. See, and Song Lin
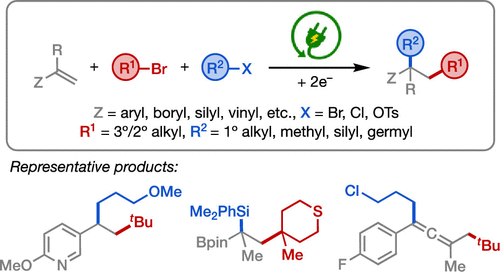
Three-Component Cross-Electrophile Coupling: Regioselective Electrochemical Dialkylation of Alkenes
The cross-electrophile dialkylation of alkenes enables the formation of two C(sp3)–C(sp3)
bonds from readily available starting materials in a single transformation, thereby
providing a modular and expedient approach to building structural complexity in organic
synthesis. Herein, we exploit the disparate electronic and steric properties of alkyl
halides with varying degrees of substitution to accomplish their selective activation
and addition to alkenes under electrochemical conditions. This method enables regioselective
dialkylation of alkenes without the use of a transition-metal catalyst and provides
access to a diverse range of synthetically useful compounds.
Complex molecule synthesis by electrocatalytic decarboxylative cross-coupling
Nature
Benxiang Zhang, Jiayan He, Yang Gao, Laura Levy, Martins S. Oderinde, Maximilian D. Palkowitz, T. G. Murali Dhar, Michael D. Mandler, Michael R. Collins, Daniel C. Schmitt, Philippe N. Bolduc, TeYu Chen, Sebastian Clementson, Nadia Nasser Petersen, Gabriele Laudadio, Cheng Bi, Yu Kawamata & Phil S. Baran
Complex molecule synthesis by electrocatalytic decarboxylative cross-coupling
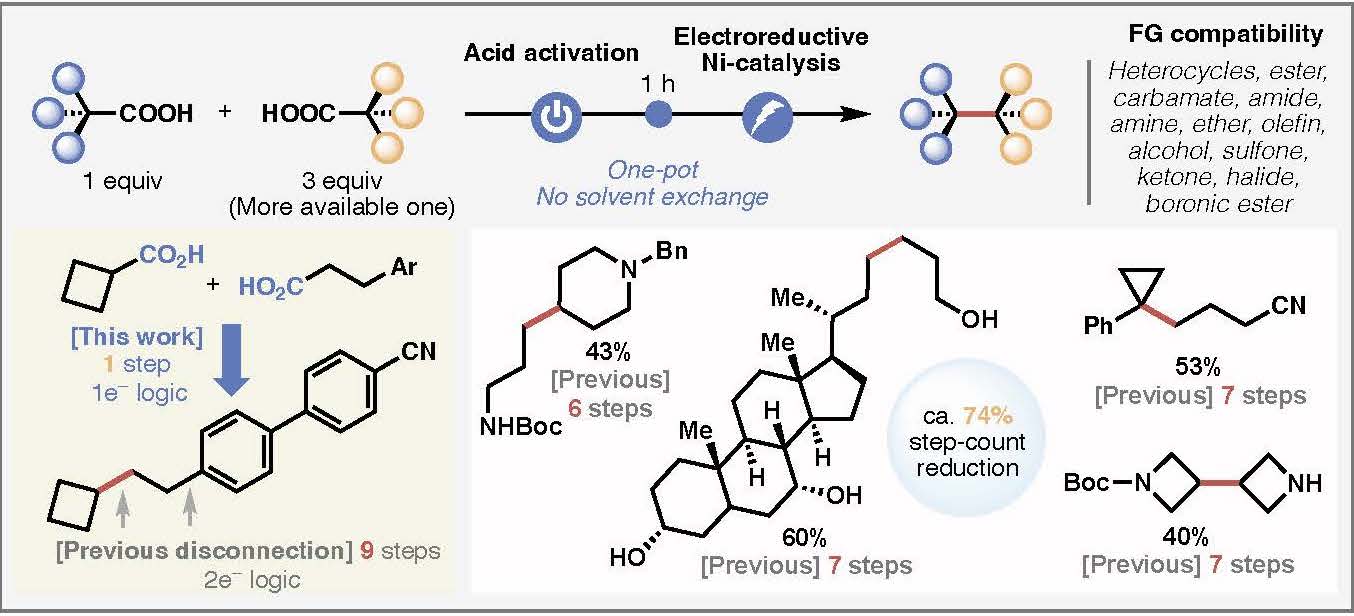
Modern retrosynthetic analysis in organic chemistry is based on the principle of polar relationships between functional groups to guide the design of synthetic routes.1 This method, termed polar retrosynthetic analysis, assigns partial positive (electrophilic) or negative (nucleophilic) charges to constituent functional groups in a complex molecules followed by disconnecting bonds between opposing charges.2–4 While this approach forms the basis of undergraduate curriculum in organic chemistry5 and strategic applications of most synthetic methods,6 their implementation often requires a long list of ancillary considerations to mitigate chemoselectivity and oxidation state issues involving protecting groups and precise reaction choreography.3,4,7 Here we report a radical-based Ni/Ag-electrocatalytic cross coupling of a-substituted carboxylic acids thereby enabling an intuitive and modular approach to accessing complex molecular architectures. This new method relies on a key silver additive that forms an active Ag-nanoparticle coated electrode surface8,9 in situ along with carefully chosen ligands that modulate the reactivity of Ni. Through judicious choice of conditions and ligands, the cross-couplings can be rendered highly diastereoselective. To demonstrate the simplifying power of these reactions, concise syntheses of 14 natural products and two medicinally relevant molecules were completed.
Scalable Electrochemical Decarboxylative Olefination Driven by Alternating Polarity
Angewandte Chemie
Benxiang Zhang, Jiayan He, Yang Gao, Laura Levy, Martins S. Oderinde, Maximilian D. Palkowitz, T. G. Murali Dhar, Michael D. Mandler, Michael R. Collins, Daniel C. Schmitt, Philippe N. Bolduc, TeYu Chen, Sebastian Clementson, Nadia Nasser Petersen, Gabriele Laudadio, Cheng Bi, Yu Kawamata & Phil S. Baran
Scalable Electrochemical Decarboxylative Olefination Driven by Alternating Polarity
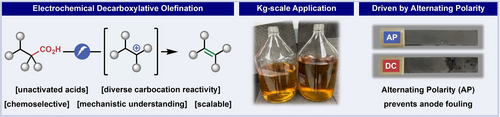
A mild, scalable (kg) metal-free electrochemical decarboxylation of alkyl carboxylic acids to olefins is disclosed. Numerous applications are presented wherein this transformation can simplify alkene synthesis and provide alternative synthetic access to valuable olefins from simple carboxylic acid feedstocks. This robust method relies on alternating polarity to maintain the quality of the electrode surface and local pH, providing a deeper understanding of the Hofer-Moest process with unprecedented chemoselectivity.
Mechanistic Insights into Electrocatalytic Carbon−Bromine Bond Cleavage in Polybrominated Phenols
JPCC
Eric C. R. McKenzie, Seyyedamirhossein Hosseini, Mayank Tanwar, Matthew Neurock, Shelley D. Minteer, and Stephen C. Jacobson
Mechanistic Insights into Electrocatalytic Carbon−Bromine Bond Cleavage in Polybrominated Phenols
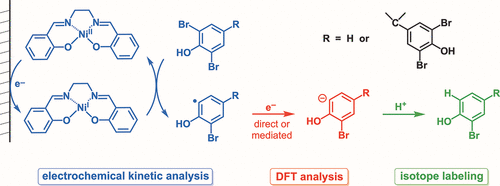
Carbon−halogen bond cleavage has been studied extensively for many years as a simple electrosynthesis step in the formation of more complex natural products. Reduction of halogenated phenols has received less attention, in part, due to the lowered faradaic efficiency resulting from the competing hydrogen evolution reaction. Herein, we report the electroreduction of a series of brominated phenols through a homogeneous electrocatalytic (EC′) mechanism. Beginning with the structurally simple 2-bromophenol, we use foot-of-the-wave analysis to determine optimal catalysts. Nickel(II) salen requires the lowest overpotential for C−Br reduction and was used across all substrates. Chronoamperometric studies and density functional theory calculations were carried out to contribute to our understanding of the reduction mechanism. Next, the more complex 2,6-dibromophenol and tetrabromobisphenol-A are studied by means of cyclic voltammetry, chronoamperometry, and density functional theory. Through analysis of molecular orbitals diagrams, the more complex brominated phenols are found to undergo sequential carbon−bromine bond reduction, wherein the electrogenerated radical species accepts a second electron to form a carbanion before second carbon−bromine bond cleavage occurs.
Unraveling Hydrogen Atom Transfer Mechanisms with Voltammetry: Oxidative Formation and Reactivity of Cobalt Hydride
JACS
Dylan G. Boucher, Andrew D. Pendergast, Xiangyu Wu, Zachary A. Nguyen, Rohit G. Jadhav, Song Lin, Henry S. White, and Shelley D. Minteer
Unraveling Hydrogen Atom Transfer Mechanisms with Voltammetry: Oxidative Formation and Reactivity of Cobalt Hydride
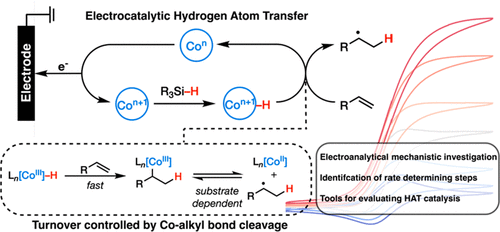
The utility of transition metal hydride catalyzed hydrogen atom transfer (MHAT) has been widely demonstrated in organic transformations such as alkene isomerization and hydrofunctionalization reactions. However, the highly reactive nature of the hydride and radical intermediates has hindered mechanistic insight into this pivotal reaction. Recent advances in electrochemical MHAT have opened up the possibility for new analytical approaches for mechanistic diagnosis. Here, we report a voltammetric interrogation of Co-based MHAT reactivity, describing in detail the oxidative formation and reactivity of the key Co–H intermediate and its reaction with aryl alkenes. Insights from cyclic voltammetry and finite element simulations help elucidate the rate-limiting step as metal hydride formation, which we show to be widely tunable based on ligand design. Voltammetry is also suggestive of the formation of Co–alkyl intermediates and a dynamic equilibrium with the reactive neutral radical. These mechanistic studies provide information for the design of future hydrofunctionalization reactions, such as catalyst and silane choice, the relative stability of metal–alkyl species, and how hydrofunctionalization reactions utilize Co–alkyl intermediates. In summary, these studies establish an important template for studying MHAT reactions from the perspective of electrochemical kinetic frameworks.
Improving the Mg Sacrificial Anode in Tetrahydrofuran for Synthetic Electrochemistry by Tailoring Electrolyte Composition
JACS Au
Wendy Zhang, Chaoxuan Gu, Yi Wang, Skyler D. Ware, Lingxiang Lu, Song Lin, Yue Qi, and Kimberly A. See
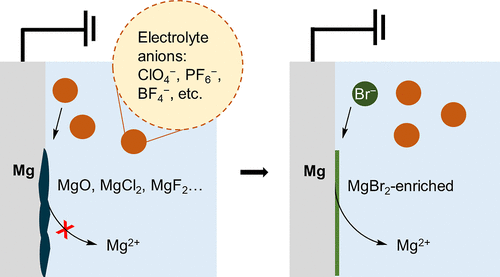
Improving the Mg Sacrificial Anode in Tetrahydrofuran for Synthetic Electrochemistry by Tailoring Electrolyte Composition
Mg0 is commonly used as a sacrificial anode in reductive electrosynthesis. While numerous
methodologies using a Mg sacrificial anode have been successfully developed, the optimization
of the electrochemistry at the anode, i.e., Mg stripping, remains empirical. In practice,
electrolytes and organic substrates often passivate the Mg electrode surface, which
leads to high overall cell potential causing poor energy efficiency and limiting reaction
scale-up. In this study, we seek to understand and manipulate the Mg metal interfaces
for a more effective counter electrode in tetrahydrofuran. Our results suggest that
the ionic interactions between the cation and the anion of a supporting electrolyte
can influence the electrical double layer, which impacts the Mg stripping efficiency.
We find halide salt additives can prevent passivation on the Mg electrode by influencing
the composition of the solid electrolyte interphase. This study demonstrates that,
by tailoring the electrolyte composition, we can modify the Mg stripping process and
enable a streamlined optimization process for the development of new electrosynthetic
methodologies.
Bioelectrocatalytic Synthesis: Concepts and Applications
Angewandte Chemie
Dylan Boucher, Emily Carroll, Zachary Nguyen, Rohit Jadhav, Olja Simoska, Kevin Beaver, Shelley D. Minteer
Bioelectrocatalytic Synthesis: Concepts and Applications
Bioelectrocatalytic synthesis is the conversion of electrical energy into value-added chemicals via a biocatalyst. Bioelectrosynthetic methods utilize the specificity and selectivity of biocatalytic enzymatic or microbial species to carry out chemical redox transformations while utilizing electricity as a stoichiometric redox equivalent. As a merging of biocatalysis and electrocatalysis, these methods directly address challenges in green and sustainable synthesis of pharmaceuticals, commodity chemicals, fuels, feedstocks and fertilizers. Despite the rising importance of bioelectrochemical transformations across these industries, there remains a high barrier for adoption due to the specialized experimental setups and domain knowledge for bioelectrocatalysis. This review aims to introduce the key concepts and design features of bioelectrosynthetic systems. A tutorial on the methods of biocatalyst utilization and the setup of bioelectrosynthetic cells is provided, as well as an overview of the analytical methods used for assessing bioelectrocatalysts. Key studies illustrating the vital applications of bioelectrosynthesis are outlined, such as ammonia production, small-molecule synthesis, and multi-carbon product formation. Finally, we address future directions for both microbial and enzymatic electrosynthetic methods. In summary, this review provides a critically necessary introduction to the field and a collection of resources for the non-specialist interested in pursuing a research program in bioelectrosynthesis.
Electrochemical Preparation of Sm(II) Reagent Facilitated by Weakly Coordinating Anions
Chemistry—A European Journal
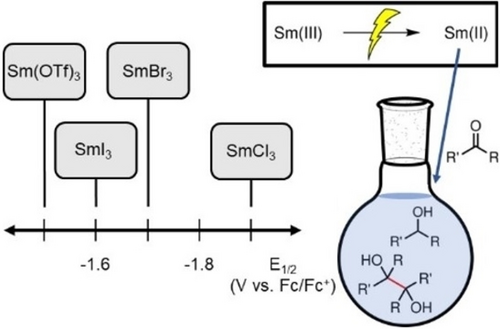
Skyler D. Ware, Wendy Zhang, David J. Charboneau, Channing K. Klein, Sarah E. Reisman, Kimberly A. See
Electrochemical Preparation of Sm(II) Reagent Facilitated by Weakly Coordinating Anions
Samarium diiodide (SmI2) is widely used as a strong one-electron reducing agent and is often employed to form C−C bonds in complex systems. Despite their utility, SmI2 and related salts suffer from several drawbacks that render the use of Sm reducing agents in large-scale synthesis impractical. Here, we report factors influencing the electrochemical reduction of Sm(III) to Sm(II), towards the goal of electrocatalytic Sm(III) reduction. We probe the effect of supporting electrolyte, electrode material, and Sm precursor on Sm(II)/(III) redox and on the reducing power of the Sm species. We find that the coordination strength of the counteranion of the Sm salt affects the reversibility and redox potential of the Sm(II)/(III) couple and establish that the counteranion primarily determines the reducibility of Sm(III). Electrochemically generated SmI2 performs similarly to commercial SmI2 solutions in a proof-of-concept reaction. The results will provide fundamental insight to facilitate the development of Sm-electrocatalytic reactions.
A Light-Promoted Innate Trifluoromethylation of Pyridones and Related N-Heteroarenes
Organic Letters
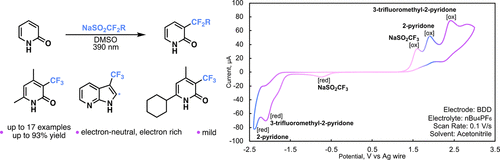
Ashley Dang-Nguyen, Kristine C. Legaspi, Connor T. McCarty, Diane K. Smith, and Jeffrey Gustafson
A Light-Promoted Innate Trifluoromethylation of Pyridones and Related N-Heteroarenes
We report a practical, light-mediated perfluoroalkylation using Langlois’ reagent
(sodium trifluoromethylsulfinate) that proceeds in the absence of any photocatalyst
or additives. This method has allowed for the facile functionalization of pyridones
and related N-heteroarenes such as azaindole. This protocol is operationally simple,
uses readily available materials, and is tolerable for electron-neutral and -rich
functional pyridones. Cyclic voltammetry was utilized as a mechanistic probe, and
preliminary data suggest the reaction may involve an electrophilic radical mechanism.
An electroaffinity labelling platform for chemoproteomic-based target identification
Nature Chemistry
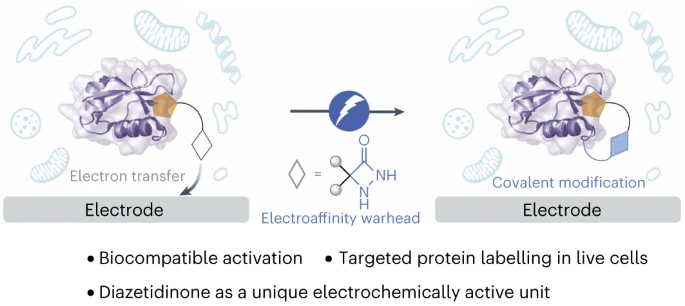
Yu Kawamata, Keun Ah Ryu, Gary N. Hermann, Alexander Sandahl, Julien C. Vantourout, Aleksandra K. Olow, La-Tonya A. Adams, Eva Rivera-Chao, Lee R. Roberts, Samer Gnaim, Molhm Nassir, Rob C. Oslund, Olugbeminiyi O. Fadeyi & Phil S. Baran
An electroaffinity labelling platform for chemoproteomic-based target identification
Target identification involves deconvoluting the protein target of a pharmacologically active, small-molecule ligand, a process that is critical for early drug discovery yet technically challenging. Photoaffinity labelling strategies have become the benchmark for small-molecule target deconvolution, but covalent protein capture requires the use of high-energy ultraviolet light, which can complicate downstream target identification. Thus, there is a strong demand for alternative technologies that allow for controlled activation of chemical probes to covalently label their protein target. Here we introduce an electroaffinity labelling platform that leverages the use of a small, redox-active diazetidinone functional group to enable chemoproteomic-based target identification of pharmacophores within live cell environments. The underlying discovery to enable this platform is that the diazetidinone can be electrochemically oxidized to reveal a reactive intermediate useful for covalent modification of proteins. This work demonstrates the electrochemical platform to be a functional tool for drug-target identification.
Anodic Cyclizations and Umpolung Reactions Involving Imines
Organic Letters
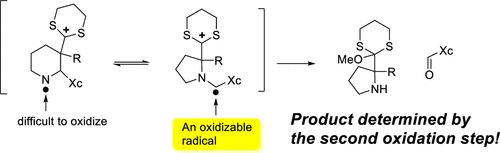
Zach Medcalf, Essence G. Redd, Jaemyeong Oh, Chang Ji, and Kevin D. Moeller
Anodic Cyclizations and Umpolung Reactions Involving Imines
Recent discoveries that anodic cyclization reactions rely heavily on the success of a second electron oxidation downstream of the cyclization suggest that this second electron oxidation step can be used to channel a reaction down new synthetic pathways. Here we describe one such application that reverses the normal reactivity of an imine group and sets the stage for the asymmetric synthesis of cyclic amines by anodic cyclization.
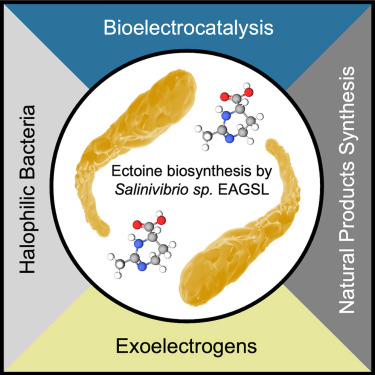
Salinivibrio sp. EAGSL as a halophilic and ectoine-producing bacteria for broad microbial electrochemistry applications
Cell Reports Physical Science
Isaac P.A. Guynn, Kevin Beaver, Erin M. Gaffney, Ana Bonizol Zani, Ashwini Dantanarayana, Shelley D. Minteer
Salinivibrio sp. EAGSL as a halophilic and ectoine-producing bacteria for broad microbial electrochemistry applications
Salinivibrio sp. EAGSL (S. EAGSL) is an extremophile that was isolated from the Great Salt Lake (UT, USA) in 2017, and this strain has since been the focus of promising research in the field of microbial electrochemistry. Namely, S. EAGSL is an organism with both halotolerance and electroactivity, giving this microbe the unique ability to bridge the gap between power output and halotolerance in microbial fuel cells. While studying the genome, a biosynthetic gene for ectoine was identified. Ectoine is an osmolyte that is deemed a value-added chemical due to its ability to stabilize proteins and other biomolecules in varying conditions, proving its importance for the biochemical and cosmetic industries. Other halophilic bacteria, including Halomonas elongata, have been previously used for industrial production of ectoine. Herein, we evaluate the ectoine production from S. EAGSL, demonstrate proof of concept for S. EAGSL-based microbial fuel cells, and offer discussion for future electrosynthesis applications.
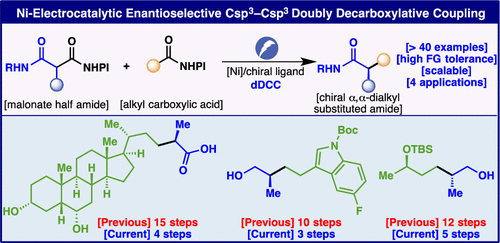
Ni-Electrocatalytic Enantioselective Doubly Decarboxylative C(sp3)–C(sp3) Cross Coupling
JACS
Yang Gao, Benxiang Zhang, Jiayan He, and Phil S. Baran
Ni-Electrocatalytic Enantioselective Doubly Decarboxylative C(sp3)–C(sp3) Cross Coupling
The first examples of enantioselective doubly decarboxylative cross coupling are disclosed. Malonate half amides are smoothly coupled to a variety of primary carboxylic acids after formation of the corresponding redox-active esters under Ni-electrocatalytic conditions using a new chiral ligand based on PyBox, resulting in amides with α-alkylated stereocenters. The scope of the reaction is broad, tolerating numerous functional groups, and uniformly proceeds with high ee. Finally, the potential utility of this enantioselective radical–radical reductive cross coupling to simplify synthesis is demonstrated with numerous case studies.
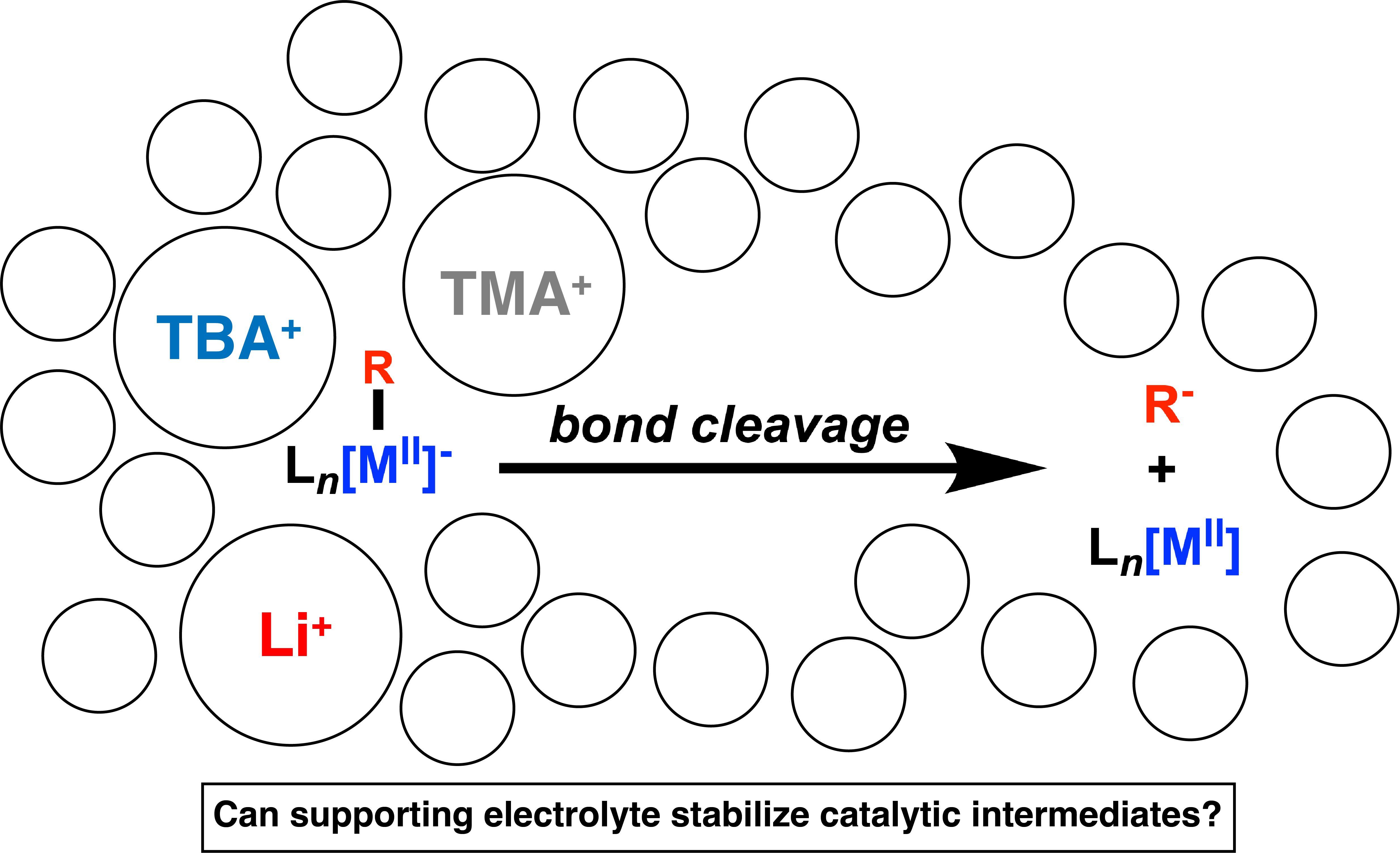
Exploring Electrolyte Effects on Metal-Alkyl Bond Stability: Impact and Implications for Electrosynthesis
Faraday Discussions
Dylan Boucher, Zachary A. Nguyen and Shelley Minteer
Exploring Electrolyte Effects on Metal-Alkyl Bond Stability: Impact and Implications for Electrosynthesis
Transition metal catalysis hinges on the formation of metal-carbon bonds during catalytic cycles. The stability and reactivity of these bonds are what determine product chemo-, regio-, and enantioselectivity. The advent of electrosynthetic methodologies has placed the current understanding of these metal-alkyl bonds into a new environment of charged species and electrochemically induced reactivity. In this paper, we explore the often neglected impact of supporting electrolyte on homogeneous electrocatalytic mechanisms using the catalytic reduction of benzyl chlorides via Co and Fe tetraphenylprophyrins as a model reaction. The mechanism of this reaction is confirmed to proceed through the formation of the metal-alkyl intermediates. Critically, the stability of these intermediates, in both the Co and Fe systems, is found to be affected by the hydrodynamic radius of the supporting electrolyte, leading to differences in electrolyte-solvent shell. These studies provide important information for the design of electrosynthetic reactions, and provide a starting point for the rational design of functional supporting electrolytes.
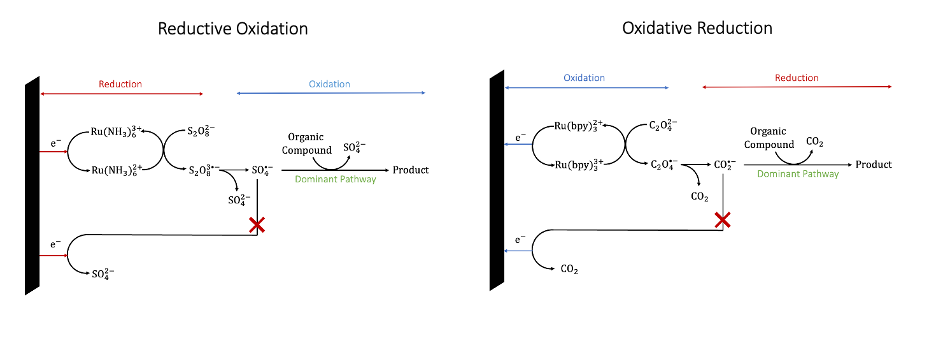
Electroorganic Synthesis in Aqueous Solution via Generation of Strongly Oxidizing and Reducing Intermediates
Faraday Discussions
 Seyyedamirhossein Hosseini, Joshua A Beeler, Melanie S Sanford and Henry S White
Seyyedamirhossein Hosseini, Joshua A Beeler, Melanie S Sanford and Henry S White
Electroorganic Synthesis in Aqueous Solution via Generation of Strongly Oxidizing and Reducing Intermediates
Although water is the ideal green solvent for organic electrosynthesis, its electrochemical window is narrow with respect to organic solvents such as acetonitrile and N,N-dimethylformamide. As such, many electroorganic processes display poor yield and selectivity in water. This work demonstrates the utility of novel synthetic strategies referred to as reductive oxidation and oxidative reduction to carry out electroorganic reactions otherwise not possible in water.
Overcoming the limitations of Kolbe coupling with waveform-controlled electrosynthesis
Science
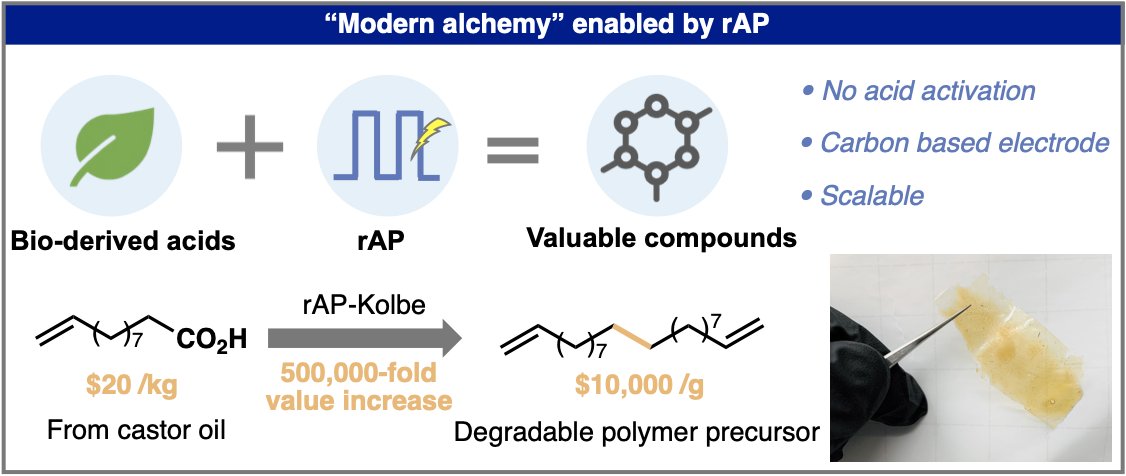
Yuta Hioki, Matteo Costantini, Jeremy Griffin, Kaid C. Harper, Melania Prado Merini, Benedikt Nissl, Yu Kawamata, Phil S. Baran
Overcoming the limitations of Kolbe coupling with waveform-controlled electrosynthesis
The Kolbe reaction forms carbon-carbon bonds through electrochemical decarboxylative coupling. Despite more than a century of study, the reaction has seen limited applications owing to extremely poor chemoselectivity and reliance on precious metal electrodes. In this work, we present a simple solution to this long-standing challenge: Switching the potential waveform from classical direct current to rapid alternating polarity renders various functional groups compatible and enables the reaction on sustainable carbon-based electrodes (amorphous carbon). This breakthrough enabled access to valuable molecules that range from useful unnatural amino acids to promising polymer building blocks from readily available carboxylic acids, including biomass-derived acids. Preliminary mechanistic studies implicate the role of waveform in modulating the local pH around the electrodes and the crucial role of acetone as an unconventional reaction solvent for Kolbe reaction.
Interrogating the Mechanistic Features of Ni(I)-Mediated Aryl Iodide Oxidative Addition Using Electroanalytical and Statistical Modeling Techniques
JACS
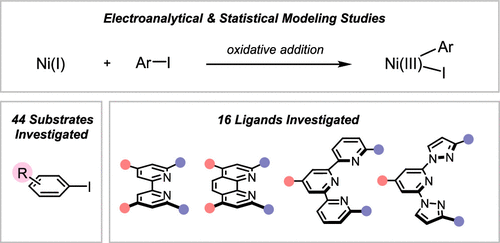
Tianhua Tang, Avijit Hazra, Daniel S. Min, Wendy L. Williams, Eli Jones, Abigail G. Doyle, and Matthew S. Sigman
Interrogating the Mechanistic Features of Ni(I)-Mediated Aryl Iodide Oxidative Addition Using Electroanalytical and Statistical Modeling Techniques
The oxidative addition of Ni(I) to aryl iodide is a crucial step in many catalytic processes, but the in-depth mechanistic understanding of this fundamental process is still lacking. We have applied electroanalytical and statistical modeling studies to understand this important mechanistic event. Our analysis led to a classification of the oxidative addition mechanism, either through a three-centered concerted or a halogen atom abstraction mechanism based on the ligand type.
Three-Stage Conversion of Chemically Inert n-Heptane to α-Hydrazino Aldehyde Based on Bioelectrocatalytic C–H Bond Oxyfunctionalization
ACS Catalysis
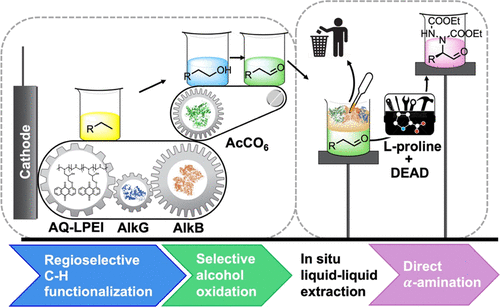 N. Samali Weliwatte, Hui Chen, Tianhua Tang, and Shelley D. Minteer
N. Samali Weliwatte, Hui Chen, Tianhua Tang, and Shelley D. Minteer
Three-Stage Conversion of Chemically Inert n-Heptane to α-Hydrazino Aldehyde Based on Bioelectrocatalytic C–H Bond Oxyfunctionalization
Petrochemical feedstocks are often used as the starting material for creating complex chemicals. However, this process can be challenging due to difficulties in selectively activating chemically inert C-H bonds and functionalizing them. In this work, the authors present a new method that combines a multienzyme cascade (enabled by an anthraquinone-based redox polymer) and organocatalysis to productively and selectively convert n-heptane to α-hydrazino aldehyde. The workflow demonstrated in this study creates a versatile and adaptable model for future research.
Unexpected Direct Synthesis of Tunable Redox-Active Benzil-Linked Polymers via the Benzoin Reaction
ACS Applied Polymer Materials
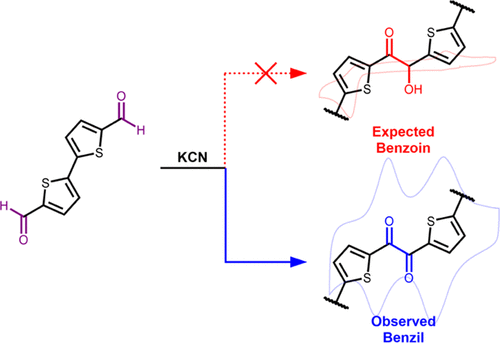
Christina Cong, Jaehwan Kim, Cara N. Gannett, Héctor D. Abruña, and Phillip J. Milner
Unexpected Direct Synthesis of Tunable Redox-Active Benzil-Linked Polymers via the Benzoin Reaction
Strategies for the sustainable synthesis of redox-active organic polymers could lead to next-generation organic electrode materials for electrochemical energy storage, electrocatalysis, and electro-swing chemical separations. Among redox-active moieties, benzils or aromatic 1,2-diones are particularly attractive due to their high theoretical gravimetric capacities and fast charge/discharge rates. Herein, we demonstrate that the cyanide-catalyzed polymerization of simple dialdehyde monomers unexpectedly leads to insoluble redox-active benzil-linked polymers instead of the expected benzoin polymers, as supported by solid-state nuclear magnetic resonance spectroscopy and electrochemical characterization. Mechanistic studies suggest that cyanide-mediated benzoin oxidation occurs by hydride transfer to the solvent and that the insolubility of the benzil-linked polymers protects them from subsequent cyanolysis. The thiophene-based polymer poly(BTDA) is an intriguing organic electrode material that demonstrates two reversible one-electron reductions with monovalent cations such as Li+ and Na+ but one two-electron reduction with divalent Mg2+. As such, the tandem benzoin-oxidation polymerization reported herein represents a sustainable method for the synthesis of highly tunable and redox-active organic materials.
Mechanistic Studies of the Electrocatalytic Carbon–Bromine Cleavage and the Hydrogen Atom Incorporation from 1, 1, 1, 3, 3, 3-Hexaflouroisopropanol
JECS
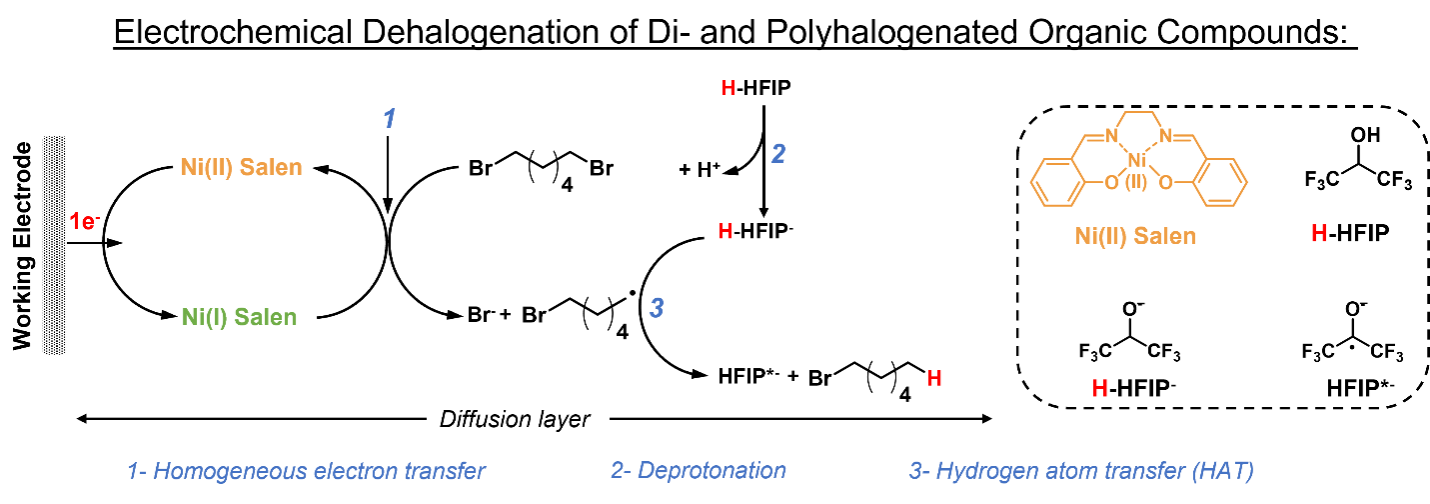
Kelly Rudman, Bishnu Thapa, Arifuzzaman Tapash, Mohammad Mubarak, Krishnan Raghavachari, Seyyedamirhossein Hosseini and Shelley D. Minteer
Mechanistic Studies of the Electrocatalytic Carbon–Bromine Cleavage and the Hydrogen Atom Incorporation from 1, 1, 1, 3, 3, 3-Hexaflouroisopropanol
Many of the environmental pollutants, such as chlorpyrifos, include multiple carbon-halogen moieties. The green approach to decontaminate these small organic molecules is to replace carbon–halogen moiety with C–H bonds. However, electrochemical cleavage of the carbon–halogen atom affords a highly unstable mono-radical that can immediately couple to form small chain polymers. These undesired byproducts then precipitate onto the working electrode and immediately shut down the electrolysis process. As a result, reactions do not reach completion and large amounts of the reactant remains unreacted. Herein, we report use of 1, 1, 1, 3, 3, 3-hexaflouroisopropanol (HFIP) as an efficient reagent to control C–H formation over radical association. Debromination of 1,6-dibromohexane was examined as a model reaction, in the presence of Ni(II) salen and HFIP as the electrocatalyst and hydrogen atom source, respectively. Our results indicate that through the use of HFIP, formation of short-chain polymers is no longer observed, and monomer formation is the dominant product.
Oxidation by Reduction: Efficient and Selective Oxidation of Alcohols by the Electrocatalytic Reduction of Peroxydisulfate
JACS
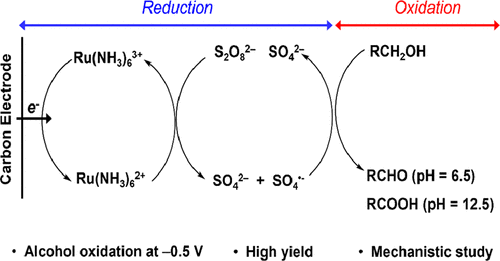
Seyyedamirhossein Hosseini, Jordyn N. Janusz, Mayank Tanwar, Andrew D. Pendergast, Matthew Neurock, and Henry S. White
Oxidation by Reduction: Efficient and Selective Oxidation of Alcohols by the Electrocatalytic Reduction of Peroxydisulfate
In this work, we have shown that SO4•– is electrocatalytically generated through the mediated reduction of S2O82– by Ru(NH3)62+ and that it can be used to carry out the reductive oxidation of benzyl alcohols to yield either benzoic acids or benzaldehydes. We believe the use of reductive oxidation for C–H activation at negative potentials provides further opportunities to carry out more complex reactions at very mild conditions such as α-carbon functionalization and cross-coupling reactions.
Investigating Oxidative Addition Mechanisms of Allylic Electrophiles with Low-Valent Ni/Co Catalysts Using Electroanalytical and Data Science Techniques
JACS
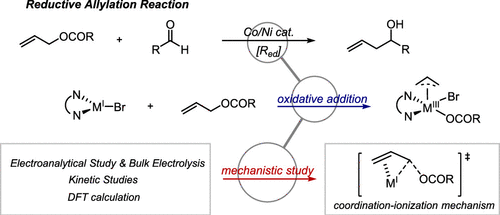 Tianhua Tang, Eli Jones, Thérèse Wild, Avijit Hazra, Shelley D. Minteer, and Matthew S. Sigman
Tianhua Tang, Eli Jones, Thérèse Wild, Avijit Hazra, Shelley D. Minteer, and Matthew S. Sigman
Investigating Oxidative Addition Mechanisms of Allylic Electrophiles with Low-Valent Ni/Co Catalysts Using Electroanalytical and Data Science Techniques
The oxidative addition mechanism between allylic electrophiles and Co(I)/Ni(I)-bipyridine complex has remained unsolved in the development of Co/Ni catalyzed reductive allylation reactions. Herein, we applied electroanalytical tools combined with physical organic studies to investigate this process. In this process, a coordination-ionization-type transition state was proposed as supported by DFT calculations.
Co-Operative Influence of O2 and H2O in the Degradation of Layered Black Arsenic
JPCC
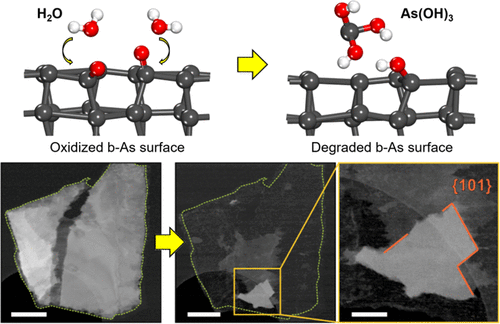 Mayank Tanwar, Sagar Udyavara, Hwanhui Yun, Supriya Ghosh, K. Andre Mkhoyan, and Matthew Neurock
Mayank Tanwar, Sagar Udyavara, Hwanhui Yun, Supriya Ghosh, K. Andre Mkhoyan, and Matthew Neurock
Co-Operative Influence of O2 and H2O in the Degradation of Layered Black Arsenic
Layered black arsenic (b-As) has recently emerged as a new anisotropic two-dimensional
(2D) semiconducting material with applications in electronic devices. Understanding
factors affecting the ambient stability of this material remains crucial for its applications.
Herein, we use first-principles density functional theory calculations to examine
the stability of the (010) and (101) surfaces of b-As in the presence of O2 and H2O.
We show that the (101) surface of b-As can easily oxidize in the presence of O2. In
the presence of moisture contained in the air, the oxidized b-As surfaces favorably
react with H2O molecules to volatilize As in the form of As(OH)3 and AsO(OH), which
results in the degradation of the b-As surface, predominantly across the (101) surface.
These predictions are in good agreement with experimental electron microscopy observations,
thus demonstrating the co-operative reactivity of O2 and H2O in the degradation of
layered b-As under ambient conditions.
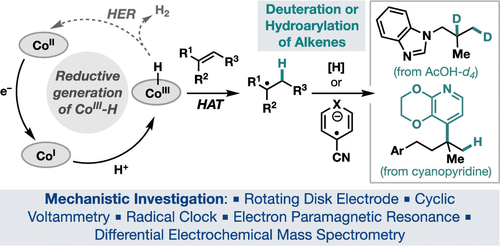
Intercepting Hydrogen Evolution with Hydrogen-Atom Transfer: Electron-Initiated Hydrofunctionalization of Alkenes
JACS
 Xiangyu Wu, Cara N. Gannett, Jinjian Liu, Rui Zeng, Luiz F. T. Novaes, Hongsen Wang,
Héctor D. Abruña, and Song Lin
Xiangyu Wu, Cara N. Gannett, Jinjian Liu, Rui Zeng, Luiz F. T. Novaes, Hongsen Wang,
Héctor D. Abruña, and Song Lin
Intercepting Hydrogen Evolution with Hydrogen-Atom Transfer: Electron-Initiated Hydrofunctionalization of Alkenes
Hydrogen-atom transfer mediated by earth-abundant transition-metal hydrides (M-Hs)
has emerged as a powerful tool in organic synthesis. Current methods to generate M-Hs
most frequently rely on oxidatively initiated hydride transfer. Herein, we report
a reductive approach to generate Co–H, which allows for canonical hydrogen evolution
reactions to be intercepted by hydrogen-atom transfer to an alkene. Electroanalytical
and spectroscopic studies provided mechanistic insights into the formation and reactivity
of Co–H, which enabled the development of two new alkene hydrofunctionalization reactions.
Overcoming Limitations in Decarboxylative Arylation via Ag–Ni Electrocatalysis
JACS
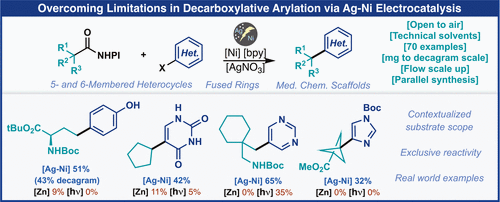 Maximilian D. Palkowitz, Gabriele Laudadio, Simon Kolb, Jin Choi, Martins S. Oderinde,
Tamara El-Hayek Ewing, Philippe N. Bolduc, TeYu Chen, Hao Zhang, Peter T. W. Cheng,
Benxiang Zhang, Michael D. Mandler, Vanna D. Blasczak, Jeremy M. Richter, Michael
R. Collins, Ryan L. Schioldager, Martin Bravo, T. G. Murali Dhar, Benjamin Vokits,
Yeheng Zhu, Pierre-Georges Echeverria, Michael A. Poss, Scott A. Shaw, Sebastian Clementson,
Nadia Nasser Petersen, Pavel K. Mykhailiuk, and Phil S. Baran
Maximilian D. Palkowitz, Gabriele Laudadio, Simon Kolb, Jin Choi, Martins S. Oderinde,
Tamara El-Hayek Ewing, Philippe N. Bolduc, TeYu Chen, Hao Zhang, Peter T. W. Cheng,
Benxiang Zhang, Michael D. Mandler, Vanna D. Blasczak, Jeremy M. Richter, Michael
R. Collins, Ryan L. Schioldager, Martin Bravo, T. G. Murali Dhar, Benjamin Vokits,
Yeheng Zhu, Pierre-Georges Echeverria, Michael A. Poss, Scott A. Shaw, Sebastian Clementson,
Nadia Nasser Petersen, Pavel K. Mykhailiuk, and Phil S. Baran
Overcoming Limitations in Decarboxylative Arylation via Ag–Ni Electrocatalysis
A useful protocol for achieving decarboxylative cross-coupling (DCC) of redox-active esters (RAE, isolated or generated in situ) and halo(hetero)arenes is reported. This pragmatically focused study employs a unique Ag–Ni electrocatalytic platform to overcome numerous limitations that have plagued this strategically powerful transformation. In its optimized form, coupling partners can be combined in a surprisingly simple way: open to the air, using technical-grade solvents, an inexpensive ligand and Ni source, and substoichiometric AgNO3, proceeding at room temperature with a simple commercial potentiostat. Most importantly, all of the results are placed into context by benchmarking with state-of-the-art methods. Applications are presented that simplify synthesis and rapidly enable access to challenging chemical space. Finally, adaptation to multiple scale regimes, ranging from parallel milligram-based synthesis to decagram recirculating flow is presented.
Decarboxylative Cross-Coupling: A Radical Tool in Medicinal Chemistry
ACS Medicinal Chemistry Letters
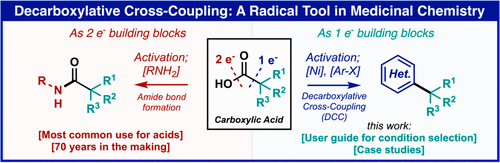
Gabriele Laudadio, Maximilian D. Palkowitz, Tamara El-Hayek Ewing, and Phil S. Baran
Decarboxylative Cross-Coupling: A Radical Tool in Medicinal Chemistry
Carboxylic acids, the most versatile and ubiquitous diversity input used in medicinal
chemistry for canonical polar bond constructions such as amide synthesis, can now
be employed in a fundamentally different category of reaction to make C–C bonds by
harnessing the power of radicals. This outlook serves as a user-guide to aid practitioners
in both the design of syntheses that leverage the simplifying power of this disconnection
and the precise tactics that can be employed to enable them. Taken together, this
emerging area holds the potential to rapidly accelerate access to chemical space of
value to modern medicinal chemistry.
Scalable, Chemoselective Nickel Electrocatalytic Sulfinylation of Aryl Halides with SO2
Angewandte Chemie
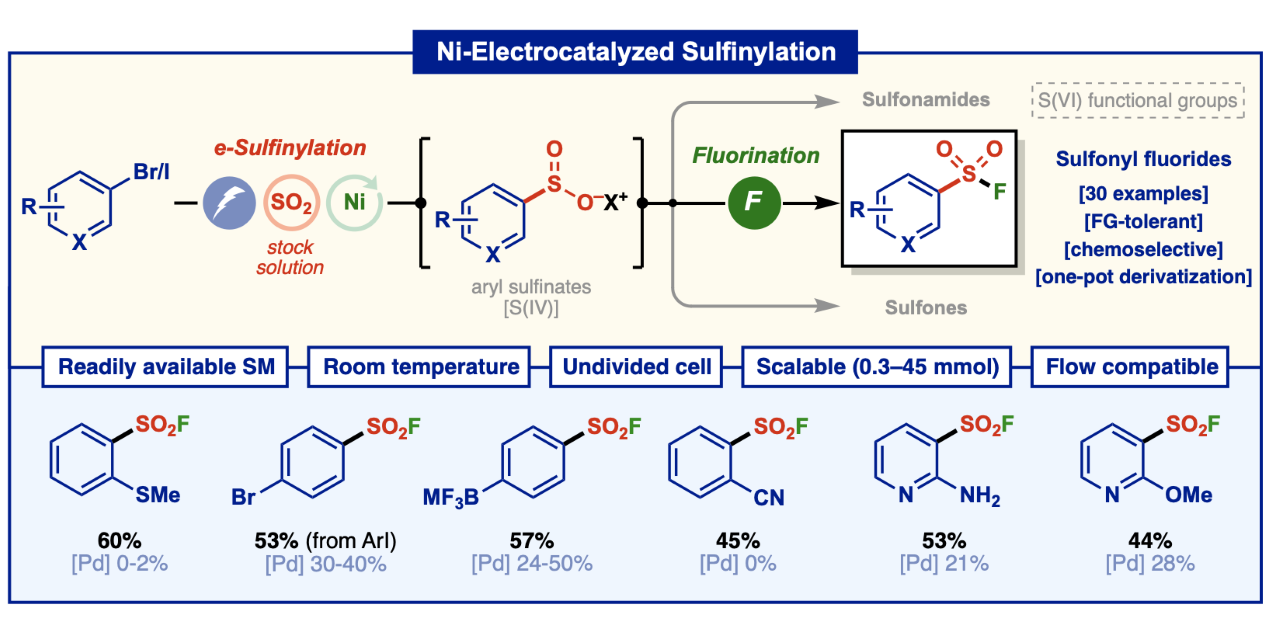
Terry Shing-Bong Lou, Yu Kawamata, Tamara Ewing, Guillermo A. Correa-Otero, Michael R. Collins, and Phil S. Baran
Scalable, Chemoselective Nickel Electrocatalytic Sulfinylation of Aryl Halides with SO2
Sulfur-containing functional groups appended to arenes are widespread in products of societal importance such as medicines, agrochemicals, and other functional materials. The most prominent precursor for such functional groups is aryl sulfinates, and direct access from readily available aryl precursors is much desired. This work presents a practical solution to prepare aryl sulfinates with inexpensive nickel catalyst under electrolytic conditions. The reaction exhibits a broad scope and can be scaled up in batch and recycle flow settings.
Cobalt-Electrocatalytic HAT for Functionalization of Unsaturated C–C Bonds
Nature
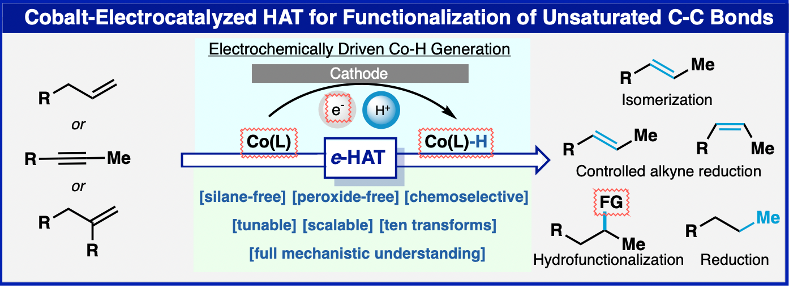
Samer Gnaim, Adriano Bauer, Hai-Jun Zhang, Longrui Chen, Cara Gannett, Christian A. Malapit, David E. Hill, David Vogt, Tianhua Tang, Ryan A. Daley, Wei Hao, Rui Zeng, Mathilde Quertenmont, Wesley D. Beck, Elya Kandahari, Julien C. Vantourout, Pierre-Georges Echeverria, Hector D. Abruna, Donna G. Blackmond, Shelley D. Minteer, Sarah E. Reisman, Matthew S. Sigman, Phil S. Baran
Cobalt-Electrocatalytic HAT for Functionalization of Unsaturated C–C Bonds
Cobalt-hydride species have found widespread uses for the derivatization and functionalization of unsaturated carbon-carbon bonds in complex molecule construction, usually via a net hydrogen atom transfer. In this work we report a new perspective on how an electrocatalytic approach inspired by decades of energy storage precedent can be leveraged in the context of efficient cobalt-hydride generation with discreet applications in modern organic synthesis. Such an approach not only offers benefits in terms of sustainability and efficiency but also enables enhanced functional group tolerance, unique chemoselectivity, and tunable reactivity. In addition, extensive mechanistic studies including computational analysis, cyclic voltammetry, Kinetic studies, and UV-vis measurements provide a comprehensive understanding of ligand-controlled Co–H generation.
Supported Pt Enabled Proton-Driven NAD(P)+ Regeneration for Biocatalytic Oxidation
ACS AMI
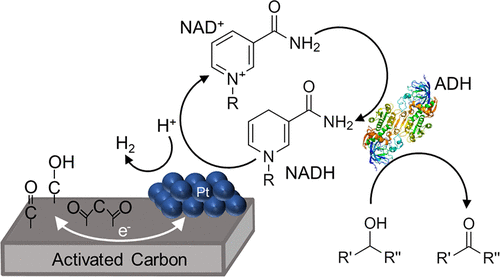
Joseph W.H. Burnett, Hui Chen, Jianwei Li, Ying Li, Shouying Huang, Jiafu Shi, Alan J. McCue, Russell F. Howe, Shelley D. Minteer, and Xiaodong Wang
Supported Pt Enabled Proton-Driven NAD(P)+ Regeneration for Biocatalytic Oxidation
The utilization of biocatalytic oxidations has evolved from the niche applications of the early 21st century to a widely recognised tool for general chemical synthesis. Heterogeneous catalysis is a form of synthetic catalysis that is directly involved in the production of 90% of chemicals globally. In this work, we demonstrate that heterogeneous catalysts (Pt/C) can promote proton-driven oxidation of NADH for selective NAD+ regeneration with concurrent H2 formation. The catalytic activity can be tuned by modifying the carbon support and altering the Pt electronic structure. The Pt/C catalysts are compatible with biocatalytic systems and can be used to support a range of biocatalytic alcohol oxidations via in situ cofactor regeneration.
Ni-Electrocatalytic C(sp3)–C(sp3) Doubly Decarboxylative Coupling
Nature
Benxiang Zhang, Yang Gao, Yuta Hioki, Martins S. Oderinde, Jennifer X. Qiao, Kevin X. Rodriguez, Hai-Jun Zhang, Yu Kawamata & Phil S. Baran
Ni-Electrocatalytic C(sp3)–C(sp3) Doubly Decarboxylative Coupling
As one of the oldest known Csp3–Csp3 bond forming reactions in synthetic chemistry, Kolbe electrolysis holds incredible promise for synthesis, yet its use has been near non-existent in mainstream organic synthesis. Here, we show how a mildly reductive Ni-electrocatalytic system can realise formal Kolbe heterocoupling of two different carboxylates via in situ generated redox-active esters (RAEs), namely doubly decarboxylative cross-coupling (dDCC). This operationally simple method, akin to the simplicity of amide bond formation, can be used to heterocouple various RAEs thereby opening up a powerful new approach to synthesis. The utility of dDCC was strategically enlisted for the synthesis of 32 known compounds to reduce overall step-counts by 73%.
Chemoselective (Hetero)Arene Electroreduction Enabled by Rapid Alternating Polarity
JACS
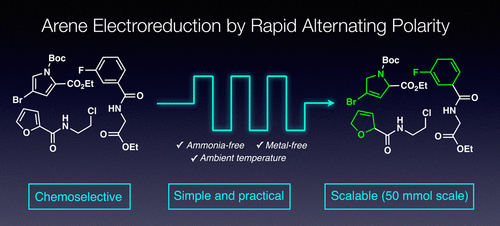
Kyohei Hayashi, Jeremy Griffin, Kaid C. Harper, Yu Kawamata, and Phil S. Baran
Chemoselective (Hetero)Arene Electroreduction Enabled by Rapid Alternating Polarity
Conventional chemical and even electrochemical Birch-type reductions suffer from a lack of chemoselectivity due to a reliance on alkali metals or harshly reducing conditions. This study reveals that applying a type of alternating current, namely rapid alternating polarity (rAP), facilitates such arene reduction at an unprecedented level of chemoselectivity. The reaction proceeds in a protic solvent and can be easily scaled up without any metal additives or stringently anhydrous conditions.Comparing Halogen Atom Abstraction Kinetics for Mn(I), Fe(I), Co(I), and Ni(I) Complexes by Combining Electroanalytical and Statistical Modeling
EurJOC
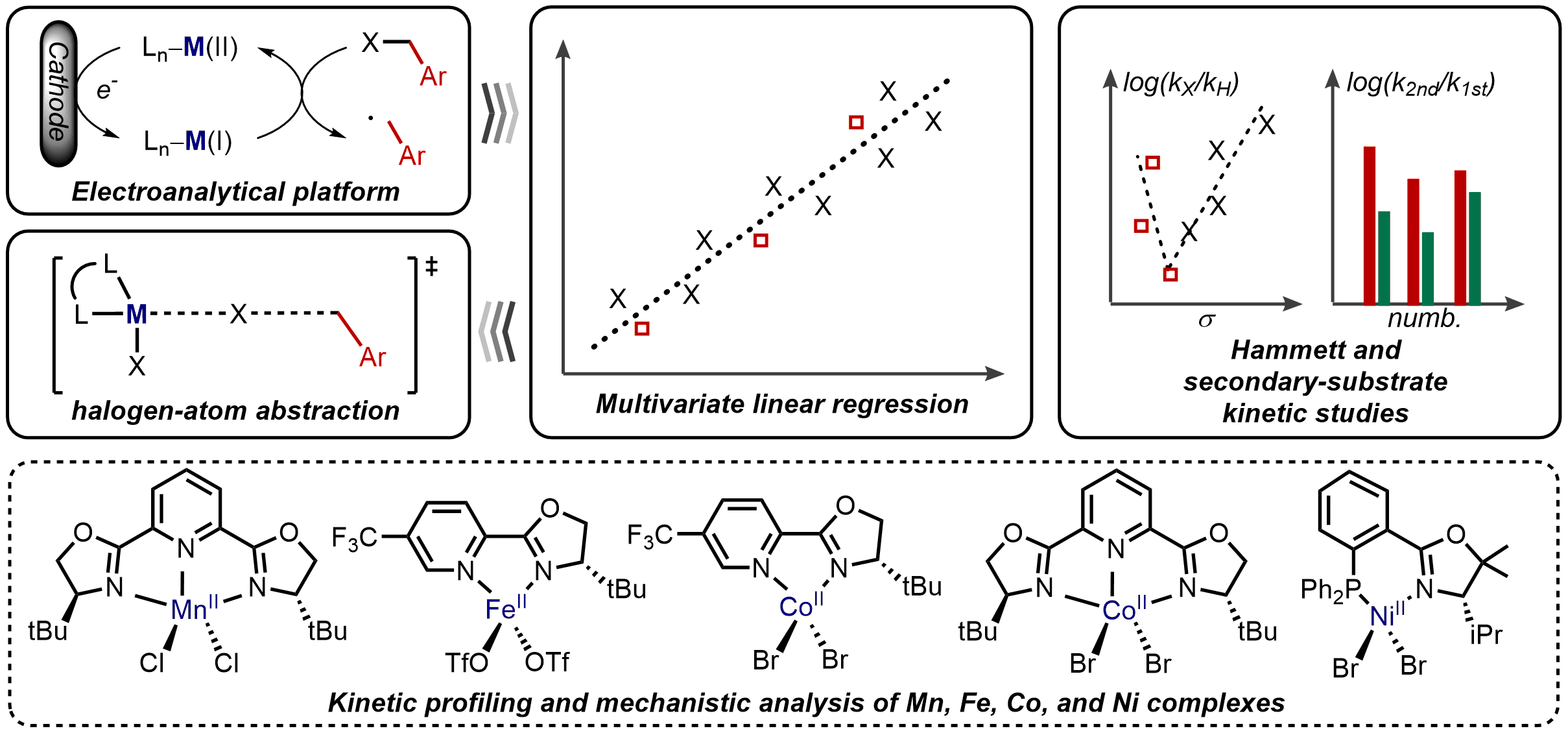
Tianhua Tang, Nathan C. Friede, Shelley D. Minteer, Matthew S. Sigman
Comparing Halogen Atom Abstraction Kinetics for Mn(I), Fe(I), Co(I), and Ni(I) Complexes by Combining Electroanalytical and Statistical Modeling
First-row transition metal complexes have garnered attention due to their ability to activate aliphatic halides for catalytic cross-coupling reactions. However, the mechanistic study for this essential activation process has been lacking. Herein, we conducted a kinetic-profiling of Mn(I)(PyBox)Cl, Fe(I)(Pyrox)OTf, Co(I)(Pyrox)Br, Co(I)(PyBox)Br, Ni(I)(Phox)Br complexes, through a combination of electroanalytical and statistical modeling. A unified halogen-atom abstraction mechanism was disclosed to account for the oxidative addition process.
Electrochemically driven cross-electrophile coupling of alkyl halides
Nature
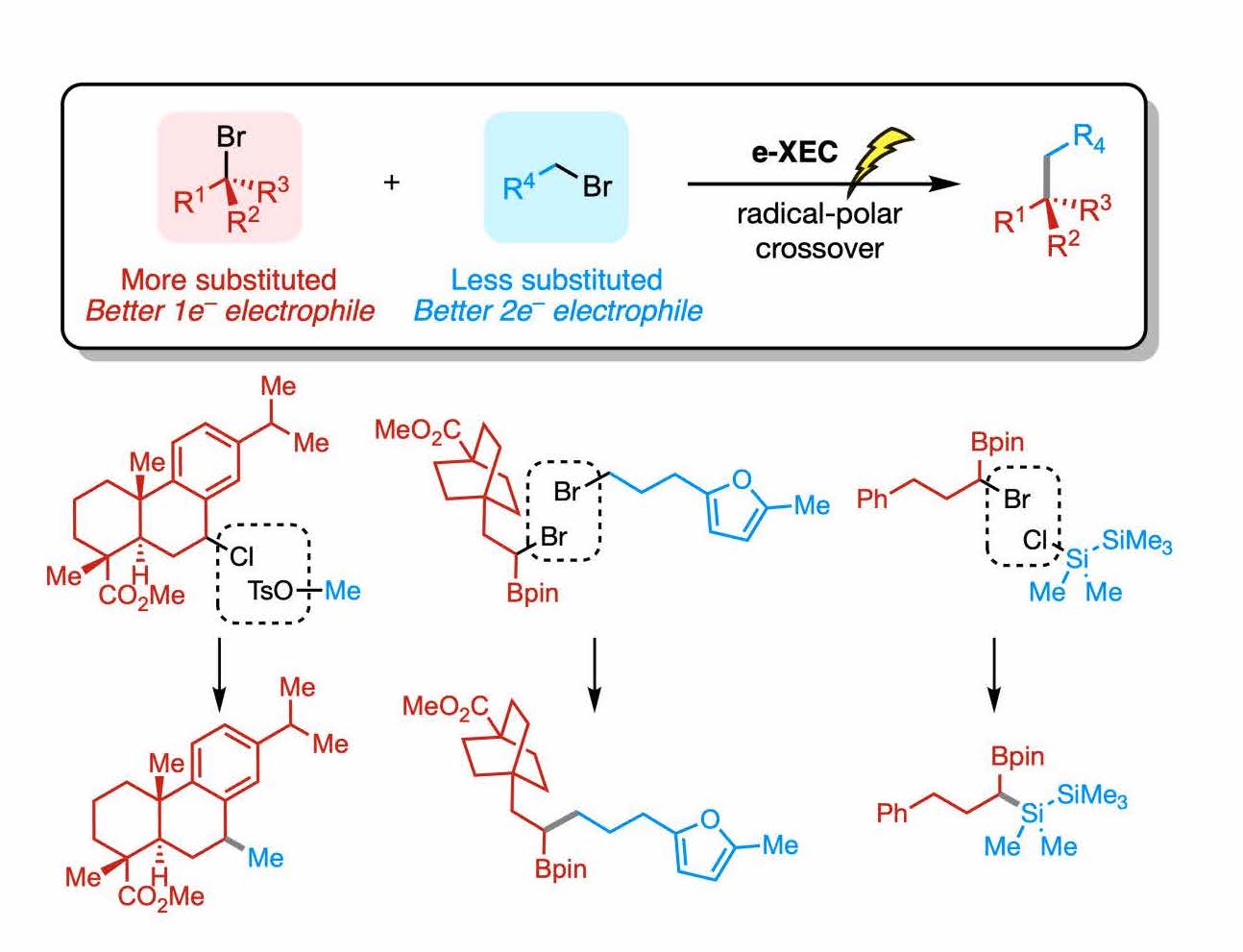
Wen Zhang, Lingxiang Lu, Wendy Zhang, Yi Wang, Skyler D. Ware, Jose Mondragon, Jonas Rein, Neil Strotman, Dan Lehnherr, Kimberly A. See & Song Lin
Electrochemically driven cross-electrophile coupling of alkyl halides
Recent research in medicinal chemistry suggests a correlation between an increase in the fraction of sp3 carbons in drug candidates with their improved success rate in clinical trials. As such, the development of robust and selective methods for the construction of C(sp3)-C(sp3) bonds remains a critical problem in modern organic chemistry. Owing to the broad availability and synthetic accessibility of alkyl halides, their direct cross coupling—commonly known as cross-electrophile electrophile-coupling (XEC)—provides a promising route toward this objective. Such transformations circumvent the preparation of carbon nucleophiles used in traditional cross-coupling reactions as well as stability and functional group tolerance issues that commonly associate with these reagents. However, achieving high selectivity in C(sp3)-C(sp3) XEC remains a largely unmet challenge. Herein, we employ electrochemistry to achieve the differential activation of alkyl halides by exploiting their disparate electronic and steric properties. Specifically, the selective cathodic reduction of a more substituted alkyl halide gives rise to a carbanion, which undergoes preferential coupling with a less substituted alkyl halide via bimolecular nucleophilic substitution (SN2) to forge a new C–C bond. This transition-metal metal-free protocol enables the efficient XEC of a variety of functionalized and unactivated alkyl electrophiles and exhibits substantially improved chemoselectivity versus existing methodologies.
Modular terpene synthesis enabled by mild electrochemical couplings
Science
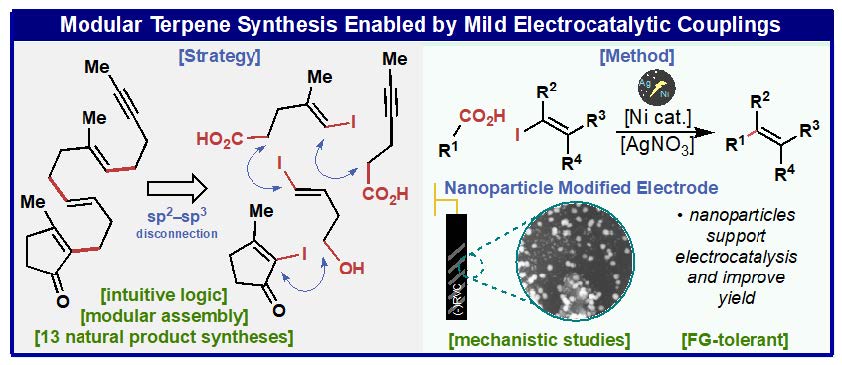
Stephen J. Harwood, Maximilian D. Palkowitz, Cara N. Gannett, Paulo Perez, Zhen Yao, Lijie Sun, Héctor D. Abruña, Scott L. Anderson, and Phil S. Baran
Modular terpene synthesis enabled by mild electrochemical couplings
This work showcases how a tactical innovation in reductive cross electrophile coupling can enable a synthetic platform to access a wide range of terpene natural products using simple modular building blocks. The key breakthrough was the discovery that silver nanoparticle functionalized electrodes help support productive catalysis for the key cross coupling reactions used to make nearly every carbon-carbon bond in the targeted molecules with minimal synthetic steps. Interdisciplinary collaborations within the center helped to demystify this discovery (Anderson and Abruna groups). These critical collaborations allowed an opportunity for the disparate worlds of material science, mechanistic electrochemistry, and total synthesis to work together to further our understanding of synthetic organic electrochemistry.
One-Pot Bioelectrocatalytic Conversion of Chemically Inert Hydrocarbons to Imines
JACS
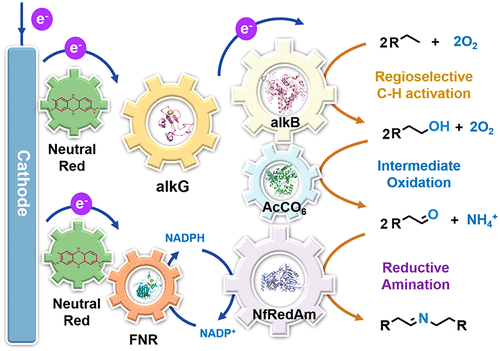
Hui Chen, Tianhua Tang, Christian A. Malapit, Yoo Seok Lee, Matthew B. Prater, N. Samali Weliwatte, and Shelley D. Minteer
One-Pot Bioelectrocatalytic Conversion of Chemically Inert Hydrocarbons to Imines
Petroleum hydrocarbons are our major energy source and an important feedstock for the chemical industry. In comparison with simply providing energy through combustion, the deep conversion of hydrocarbons to more valuable chemicals with more complicated structures for commodities and fine chemicals is of more considerable interest for the chemical industry, as that has the potential to have a substantial economic impact. In this study, our novel bioelectrocatalytic hydrocarbon deep conversion system successfully realized the challenging conversion from inert hydrocarbons to imines that would have been impossible using organic synthesis methods and provided a new methodology for the comprehensive conversion and utilization of inert hydrocarbons.
Exploring Electrochemical C(sp3)–H Oxidation for the Late-Stage Methylation of Complex Molecules
JACS
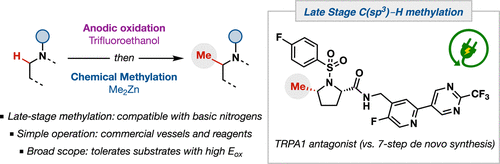
Luiz F. T. Novaes, Justin S. K. Ho, Kaining Mao, Kaida Liu, Mayank Tanwar, Matthew Neurock, Elisia Villemure, Jack A. Terrett, and Song Lin
Exploring Electrochemical C(sp3)–H Oxidation for the Late-Stage Methylation of Complex Molecules
The “magic methyl” effect, a dramatic boost in the potency of biologically active compounds from the incorporation of a single methyl group, provides a simple yet powerful strategy employed by medicinal chemists in the drug discovery process. In this work, a selective methylation protocol of complex molecules was achieved with a simple experimental setup, taking advantage of a classical electrochemical reaction, the Shono oxidation. The combination of experiments with computational studies led to a new mechanistic proposal for the Shono oxidation.
Lessons from an Array: Using An Electrode Surface to Control The Selectivity of a Solution Phase Chemical Reaction
Angewandte Chemie
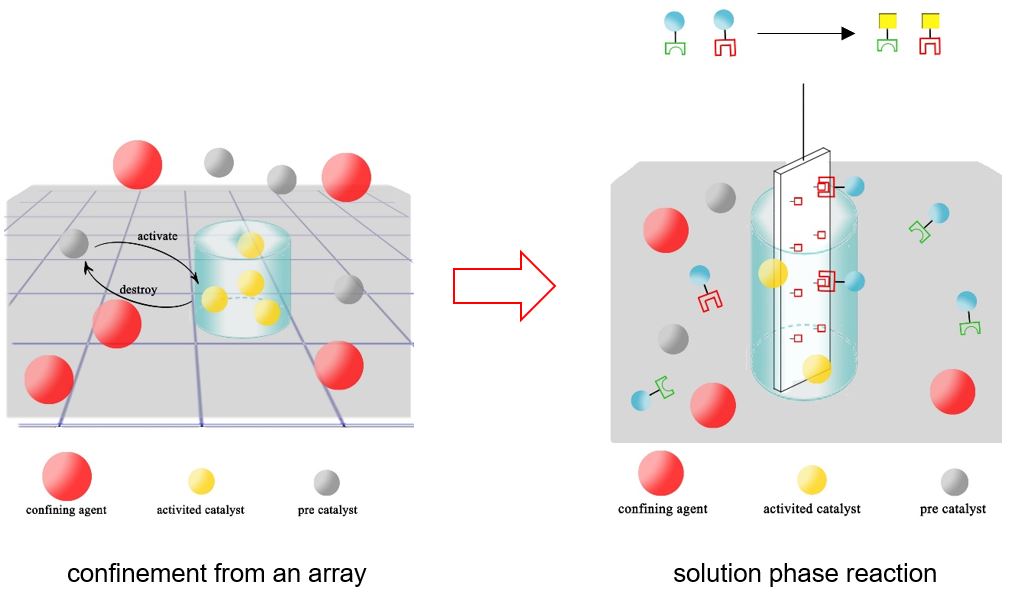
Enqi Feng, Qiwei Jing, Kevin David Moeller
Lessons from an Array: Using An Electrode Surface to Control The Selectivity of a Solution Phase Chemical Reaction
Electrochemistry offers a variety of novel means by which selectivity can be introduced into synthetic organic transformations. In the work reported, it is shown how methods used to confine chemical reactions to specific sites on a microelectrode array can also be used to confine a preparative reaction to the surface of an electrode inserted into a bulk reaction solution. In so doing, the surface of a modified electrode can be used to introduce new selectivity into a preparative reaction that is not observed in the absence of either the modified electrode surface or the effort to confine the reaction to that surface. The observed selectivity can be optimized in the same way that confinement is optimized on an array and is dependent on the nature of the functionalized surface.
Advances on the Merger of Electrochemistry and Transition Metal Catalysis for Organic Synthesis
Chemical Reviews
Christian A. Malapit, Matthew B. Prater, Jaime R. Cabrera-Pardo, Min Li, Tammy D. Pham, Timothy Patrick McFadden, Skylar Blank, and Shelley D. Minteer
Advances on the Merger of Electrochemistry and Transition Metal Catalysis for Organic Synthesis
This review summarizes the recent advances on utilizing electrochemistry and homogeneous transition metal catalysis for organic synthesis. These advancements were made possible by the increased recognition of electrochemistry as a highly effective benign reagent in electron transfer processes to generate highly reactive intermediates. This review demonstrates that electrocatalysis, through the merger of homogenous transition metal catalyst and electrochemistry, has greatly expanded the scope and improved the selectivity and reaction conditions in many important and challenging transformations.
Conjugated Microporous Polymers via Solvent-Free Ionothermal Cyclotrimerization of Methyl Ketones
Chemistry of Materials
Jaehwan Kim, Casandra M. Moisanu, Cara N. Gannett, Arjun Halder, José J. Fuentes-Rivera, Sean H. Majer, Kyle M. Lancaster, Alexander C. Forse, Héctor D. Abruña, and Phillip J. Milner
Conjugated Microporous Polymers via Solvent-Free Ionothermal Cyclotrimerization of Methyl Ketones
Conjugated microporous polymers (CMPs) are porous organic materials that display (semi)conducting behavior due to their highly π-conjugated structures. As such, they are promising next-generation materials for applications requiring both conductivity and porosity, such as supercapacitive energy storage and electrochemical sensing. However, most CMPs and related porous aromatic frameworks (PAFs) are currently prepared using expensive transition metal-based catalysts under solvothermal conditions, significantly increasing their manufacturing costs. Herein, we demonstrate that the ionothermal cyclotrimerization of methyl ketones via the aldol reaction represents a new strategy for the solvent-free synthesis of CMPs and PAFs. Specifically, we show that 1,3,5-triacetylbenzene and tetrakis(4-acetylphenyl)methane can be polymerized in molten zinc chloride to produce highly conjugated and microporous materials, as confirmed by 77 K N2 adsorption measurements in conjunction with UV–vis, Raman, and solid-state NMR spectroscopies. The CMP prepared from 1,3,5-triacetylbenzene demonstrates higher charge storage capacities (up to 172 F/g) than a commercially available supercapacitor carbon, reflecting the promise of cyclotrimerized CMPs for electrical energy storage applications.
Engineering Cyanobacterium with Transmembrane Electron Transfer Ability for Bioelectrochemical Nitrogen Fixation
ACS Catalysis
Fangyuan Dong, Yoo Seok Lee, Erin M. Gaffney, Willisa Liou, and Shelley D. Minteer
Engineering Cyanobacterium with Transmembrane Electron Transfer Ability for Bioelectrochemical Nitrogen Fixation
Cyanobacteria are excellent platforms for the synthesis of valuable compounds. The insulate outer membrane of cyanobacteria has been a limiting factor for its applications in bioelectrosynthesis. Here, an outer membrane protein was cloned and expressed in S. elongatus PCC 7942 to realize direct transmembrane electron transfer to unlock its potential in bioelectrochemical nitrogen fixation. The cellular electron transfer pathways were also briefly investigated in this work to unveil how the external electrons contribute to cyanobacterial electrosynthesis.
Capitalizing on Mediated Electrolyses for the Construction of Complex, Addressable Molecular Surfaces
JOC
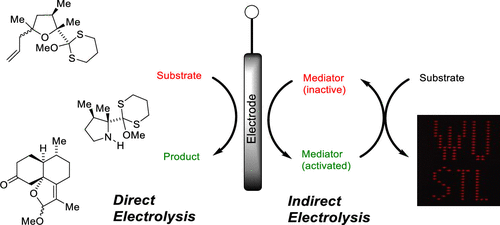
Ruby Krueger and Kevin D. Moeller
Capitalizing on Mediated Electrolyses for the Construction of Complex, Addressable Molecular Surfaces
Synthetic organic chemists are beginning to exploit electrochemical methods in increasingly creative ways. This is leading to a surge in productivity that is only now starting to take advantage of the full-potential of electrochemistry for accessing new structures in novel, more efficient ways. In this perspective, we provide insight into the potential of electrochemistry as a synthetic tool gained through studies of both direct anodic oxidation reactions and more recent indirect methods, and highlight how the development of new electrochemical methods can expand the nature of synthetic problems our community can tackle.
Chemoselective Electrosynthesis Using Rapid Alternating Polarity
JACS
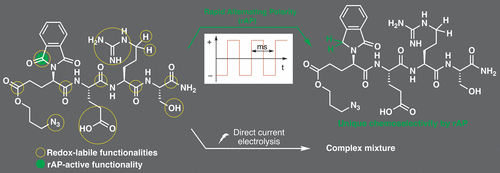
Yu Kawamata, Kyohei Hayashi, Ethan Carlson, Shobin Shaji, Dirk Waldmann, Bryan J. Simmons, Jacob T. Edwards, Christoph W. Zapf, Masato Saito, and Phil S. Baran
Chemoselective Electrosynthesis Using Rapid Alternating Polarity
Historically, electrosynthesis has been performed almost solely by using direct current (DC), whereas applying alternating current (AC) has been largely underexplored regardless of the fact that such an approach is known to change reaction outcomes considerably on an analytical scale. In this work we revealed how a square waveform–rapid alternating polarity (rAP)–enables unique chemoselectivity that cannot be achieved by conventional DC electrolysis, and demonstrated the utility of such high chemoselectivity in real-world applications such as selective manipulation of densely functionalized molecules and derivatization of PROTACs.
Chemoselective, Scalable Nickel-Electrocatalytic O-Arylation of Alcohols
Angewandte Chemie
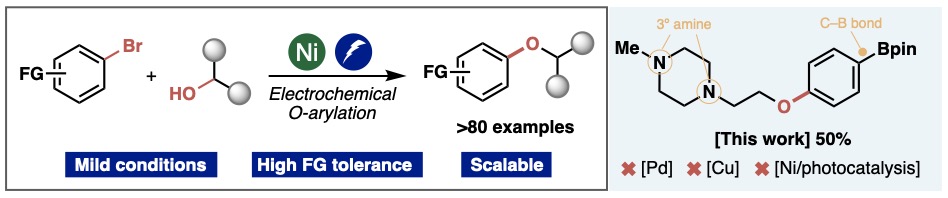
Hai-Jun Zhang, Longrui Chen, Martins S. Oderinde, Jacob T. Edwards, Yu Kawamata, and Phil S. Baran
Chemoselective, Scalable Nickel-Electrocatalytic O-Arylation of Alcohols
The formation of aryl-alkyl ether bonds through cross coupling of alcohols with aryl halides represents a complementary approach to classical SN2 methods, allowing chemists to access ethers for which conventional SN2 methods do not work well. In this work we disclose a Ni-catalyzed electrochemically driven protocol (e-etherification) to achieve this useful transformation with a broad substrate scope in an operationally simple way. This method does not require strong base, exogenous expensive transition metal catalysts (e.g. Ir, Ru, Pd), and can easily be scaled up. Interestingly, e-etherification exhibits an enhanced substrate scope over the mechanistically related photochemical variant, successfully achieving short-step syntheses of various pharmaceutically relevant ethers.
Advances in electrochemical cofactor regeneration: enzymatic and non-enzymatic approaches
Current Opinion in Biotechnology
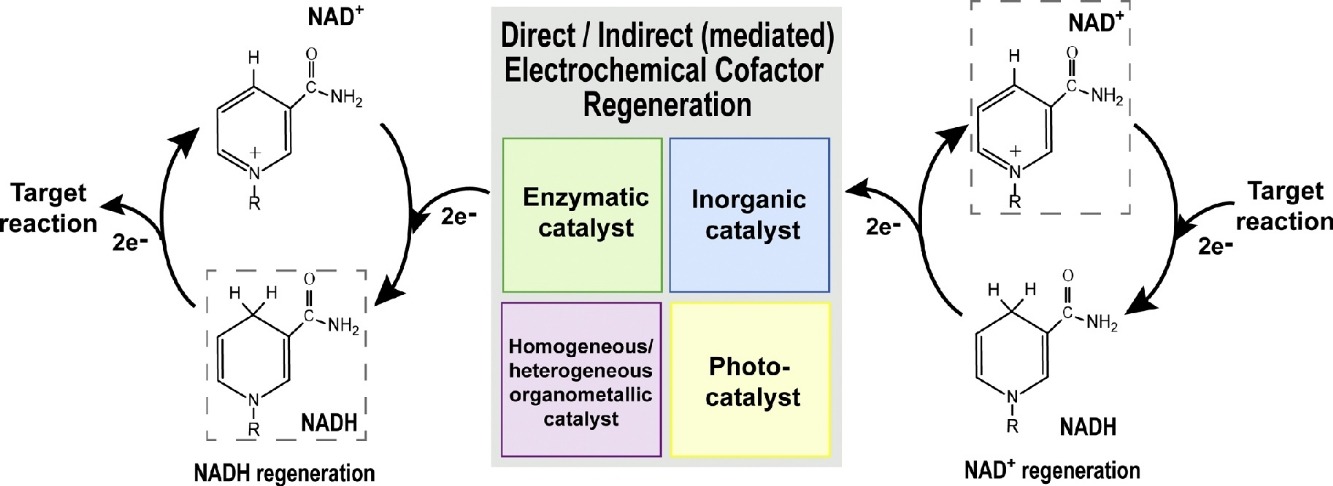
Yoo Seok Lee, Rokas Gerulskis, Shelley D. Minteer
Advances in electrochemical cofactor regeneration: enzymatic and non-enzymatic approaches
Nicotinamide adenine dinucleotide(NAD(P)H) is a metabolically interconnected redox cofactor serving as a hydride source for the majority of oxidoreductases, and consequently constituting a significant cost factor for bioprocessing. Much research has been devoted to the development of efficient, affordable, and sustainable methods for the regeneration of these cofactors through chemical, electrochemical, and photochemical approaches. However, the enzymatic approach using formate dehydrogenase is still the most abundantly employed in industrial applications, even though it suffers from system complexity and product purity issues. In this review, we summarize non-enzymatic and enzymatic electrochemical approaches for cofactor regeneration, then discuss recent developments to solve major issues. Issues discussed include Rh-catalyst mediated enzyme mutual inactivation, electron-transfer rates, catalyst sustainability, product selectivity and simplifying product purification. Recently reported remedies are discussed, such as heterogeneous metal catalysts generating H+ as the sole byproduct or high activity and stability redox-polymer immobilized enzymatic systems for sustainable organic synthesis.
Electrochemical Nozaki–Hiyama–Kishi Coupling: Scope, Applications, and Mechanism
JACS
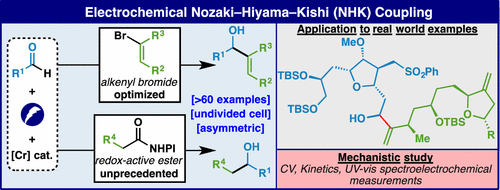
Yang Gao, David E. Hill, Wei Hao, Brendon J. McNicholas, Julien C. Vantourout, Ryan G. Hadt, Sarah E. Reisman, Donna G. Blackmond, and Phil S. Baran
Electrochemical Nozaki–Hiyama–Kishi Coupling: Scope, Applications, and Mechanism
One of the most oft-employed methods for C–C bond formation involving the coupling of vinyl-halides with aldehydes catalyzed by Ni and Cr (Nozaki–Hiyama–Kishi, NHK) has been rendered more practical using an electroreductive manifold. Although early studies pointed to the feasibility of such a process, those precedents were never applied by others due to cumbersome setups and limited scope. Here we show that a carefully optimized electroreductive procedure can enable a more sustainable approach to NHK, even in an asymmetric fashion on highly complex medicinally relevant systems. The e-NHK can even enable non-canonical substrate classes, such as redox-active esters, to participate with low loadings of Cr when conventional chemical techniques fail. A combination of detailed kinetics, cyclic voltammetry, and in situ UV–vis spectroelectrochemistry of these processes illuminates the subtle features of this mechanistically intricate process.
N-Ammonium Ylide Mediators for Electrochemical C–H Oxidation
JACS
Masato Saito, Yu Kawamata, Michael Meanwell, Rafael Navratil, Debora Chiodi, Ethan Carlson, Pengfei Hu, Longrui Chen, Sagar Udyavara, Cian Kingston, Mayank Tanwar, Sameer Tyagi, Bruce P. McKillican, Moses G. Gichinga, Michael A. Schmidt, Martin D. Eastgate, Massimiliano Lamberto, Chi He, Tianhua Tang, Christian A. Malapit, Matthew S. Sigman, Shelley D. Minteer, Matthew Neurock, and Phil S. Baran
N-Ammonium Ylide Mediators for Electrochemical C–H Oxidation
The site-specific oxidation of strong C(sp3)–H bonds is of uncontested utility in organic synthesis. From simplifying access to metabolites and late-stage diversification of lead compounds to truncating retrosynthetic plans, there is a growing need for new reagents and methods for achieving such a transformation in both academic and industrial circles. One main drawback of current chemical reagents is the lack of diversity with regard to structure and reactivity that prevents a combinatorial approach for rapid screening to be employed. In that regard, directed evolution still holds the greatest promise for achieving complex C–H oxidations in a variety of complex settings. Herein we present a rationally designed platform that provides a step toward this challenge using N-ammonium ylides as electrochemically driven oxidants for site-specific, chemoselective C(sp3)–H oxidation. By taking a first-principles approach guided by computation, these new mediators were identified and rapidly expanded into a library using ubiquitous building blocks and trivial synthesis techniques. The ylide-based approach to C–H oxidation exhibits tunable selectivity that is often exclusive to this class of oxidants and can be applied to real-world problems in the agricultural and pharmaceutical sectors.
Interfacial toughening with self-assembled monolayers enhances perovskite solar cell reliability
Science
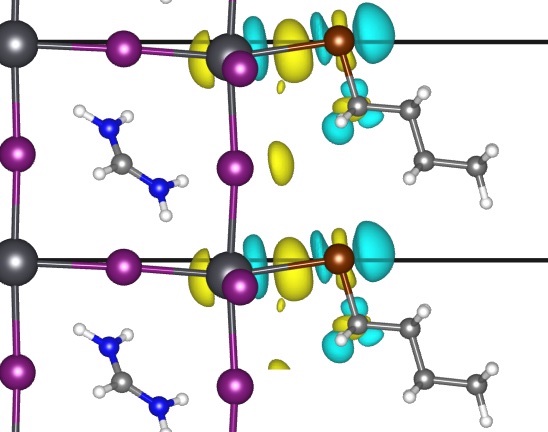
Zhenghong Dai, Srinivas K. Yadavalli, Min Chen, Ali Abbaspourtamijani, Yue Qi, Nitin P. Padture
Interfacial toughening with self-assembled monolayers enhances perovskite solar cell reliability
Iodine-terminated self-assembled monolayer (I-SAM) was used in perovskite solar cells (PSCs) to achieve a 50% increase of adhesion toughness at the interface between the electron transport layer (ETL) and the halide perovskite thin film to enhance mechanical reliability. Treatment with I-SAM also increased the power conversion efficiency from 20.2% to 21.4%, reduced hysteresis, and improved operational stability with a projected T80 (time to 80% initial efficiency retained) increasing from ~700 hours to 4000 hours under 1-sun illumination and with continuous maximum power point tracking. Operational stability–tested PSC without SAMs revealed extensive irreversible morphological degradation at the ETL/perovskite interface, including voids formation and delamination, whereas PSCs with I-SAM exhibited minimal damage accumulation. This difference was attributed to a combination of a decrease in hydroxyl groups at the interface and the higher interfacial toughness.
Effect of Pd Coordination and Isolation on the Catalytic Reduction of O2 to H2O2 over PdAu Bimetallic Nanoparticles
JACS
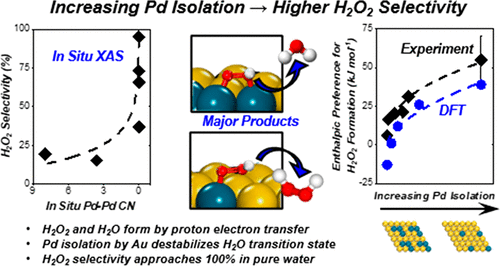
Tomas Ricciardulli, Sahithi Gorthy, Jason S. Adams, Coogan Thompson, Ayman M. Karim, Matthew Neurock, and David W. Flaherty
Effect of Pd Coordination and Isolation on the Catalytic Reduction of O2 to H2O2 over PdAu Bimetallic Nanoparticles
The direct synthesis of hydrogen peroxide (H2 + O2 → H2O2) may enable low-cost H2O2 production and reduce environmental impacts of chemical oxidations. Here, we synthesize a series of Pd1Aux nanoparticles (where 0 ≤ x ≤ 220, ∼10 nm) and show that, in pure water solvent, H2O2 selectivity increases with the Au to Pd ratio and approaches 100% for Pd1Au220. Analysis of in situ XAS and ex situ FTIR of adsorbed 12CO and 13CO show that materials with Au to Pd ratios of ∼40 and greater expose only monomeric Pd species during catalysis and that the average distance between Pd monomers increases with further dilution. Ab initio quantum chemical simulations and experimental rate measurements indicate that both H2O2 and H2O form by reduction of a common OOH* intermediate by proton–electron transfer steps mediated by water molecules over Pd and Pd1Aux nanoparticles. Measured apparent activation enthalpies and calculated activation barriers for H2O2 and H2O formation both increase as Pd is diluted by Au, even beyond the complete loss of Pd–Pd coordination. These effects impact H2O formation more significantly, indicating preferential destabilization of transition states that cleave O–O bonds reflected by increasing H2O2 selectivities (19% on Pd; 95% on PdAu220) but with only a 3-fold reduction in H2O2 formation rates. The data imply that the transition states for H2O2 and H2O formation pathways differ in their coordination to the metal surface, and such differences in site requirements require that we consider second coordination shells during the design of bimetallic catalysts.
Electrochemically driven desaturation of carbonyl compounds
Nature Chemistry
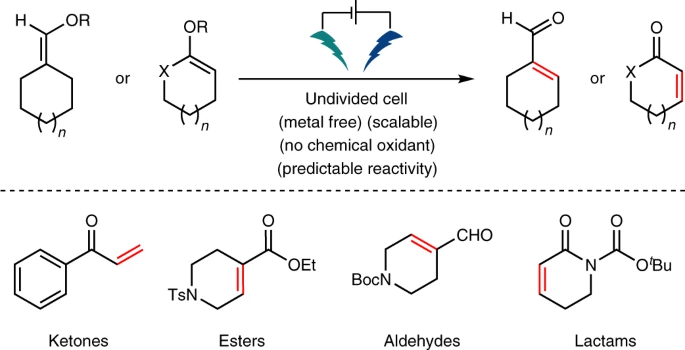
Samer Gnaim, Yusuke Takahira, Henrik R. Wilke, Zhen Yao, Jinjun Li, Dominique Delbrayelle, Pierre-Georges Echeverria, Julien C. Vantourout & Phil S. Baran
Electrochemically driven desaturation of carbonyl compounds
Electrochemical techniques have long been heralded for their innate sustainability as efficient methods to achieve redox reactions. Carbonyl desaturation, as a fundamental organic oxidation, is an oft-employed transformation to unlock adjacent reactivity through the formal removal of two hydrogen atoms. To date, the most reliable methods to achieve this seemingly trivial reaction rely on transition metals (Pd or Cu) or stoichiometric reagents based on I, Br, Se or S. Here we report an operationally simple pathway to access such structures from enol silanes and phosphates using electrons as the primary reagent. This electrochemically driven desaturation exhibits a broad scope across an array of carbonyl derivatives, is easily scalable (1–100 g) and can be predictably implemented into synthetic pathways using experimentally or computationally derived NMR shifts. Systematic comparisons to state-of-the-art techniques reveal that this method can uniquely desaturate a wide array of carbonyl groups. Mechanistic interrogation suggests a radical-based reaction pathway.
Analyzing mechanisms in Co(i) redox catalysis using a pattern recognition platform
Chemical Science
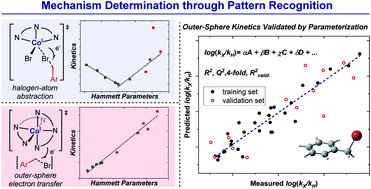 Tianhua Tang, Christopher Sandford, Shelley D. Minteer, and Matthew S. Sigman
Tianhua Tang, Christopher Sandford, Shelley D. Minteer, and Matthew S. Sigman
Analyzing mechanisms in Co(i) redox catalysis using a pattern recognition platform
Redox catalysis has been broadly utilized in electrochemical synthesis due to its kinetic advantages over direct electrolysis. The appropriate choice of redox mediator can avoid electrode passivation and overpotential, which strongly inhibit the efficient activation of substrates in electrolysis. Despite the benefits brought by redox catalysis, establishing the precise nature of substrate activation remains challenging. Herein, we determine that a Co(I) complex bearing two N,N,N-tridentate ligands acts as a competent redox catalyst for the reduction of benzyl bromide substrates. Kinetic studies combining electroanalytical techniques with multivariable linear-regression analysis were conducted, disclosing an outer-sphere electron-transfer mechanism, which occurs in concert with C–Br bond cleavage. Furthermore, we apply a pattern recognition platform to distinguish between mechanisms in the activation of benzyl bromides, found to be dependent on the ligation state of the cobalt(I) center and ligand used.
Advances in Electrochemical Modification Strategies of 5‐Hydroxymethylfurfural
ChemSusChem
Olja Simoska, Zayn Rhodes, Samali Weliwatte, Jaime R. Cabrera‐Pardo, Erin M. Gaffney, Koun Lim, Shelley D. Minteer
Advances in Electrochemical Modification Strategies of 5‐Hydroxymethylfurfural
The development of electrochemical catalytic conversion of 5‐hydroxymethylfurfural (HMF) has recently gained attention as a potentially scalable approach for both oxidation and reduction processes yielding value‐added products. While the possibility of electrocatalytic HMF transformations has been demonstrated, this growing research area is in its initial stages. Additionally, its practical applications remain limited due to low catalytic activity and product selectivity. Understanding the catalytic processes and design of electrocatalysts are important in achieving a selective and complete conversion into the desired highly valuable products. In this Minireview, an overview of the most recent status, advances, and challenges of oxidation and reduction processes of HMF was provided. Discussion and summary of voltammetric studies and important reaction factors (e. g., catalyst type, electrode material) were included. Finally, biocatalysts (e. g., enzymes, whole cells) were introduced for HMF modification, and future opportunities to combine biocatalysts with electrochemical methods for the production of high‐value chemicals from HMF were discussed.
Solvent molecules form surface redox mediators in situ and cocatalyze O2 reduction on Pd
Science
Jason S. Adams, Ashwin Chemburkar, Pranjali Priyadarshini, Tomas Ricciardulli, Yubing Lu, Vineet Maliekkal, Abinaya Sampath, Stuart Winikoff, Ayman M. Karim, Matthew Neurock, David W. Flaherty
Solvent molecules form surface redox mediators in situ and cocatalyze O2 reduction on Pd
Solvent molecules influence the reactions of molecular hydrogen and oxygen on palladium nanoparticles. Organic solvents activate to form reactive surface intermediates that mediate oxygen reduction through pathways distinct from reactions in pure water. Kinetic measurements and ab initio quantum chemical calculations indicate that methanol and water cocatalyze oxygen reduction by facilitating proton-electron transfer reactions. Methanol generates hydroxymethyl intermediates on palladium surfaces that efficiently transfer protons and electrons to oxygen to form hydrogen peroxide and formaldehyde. Formaldehyde subsequently oxidizes hydrogen to regenerate hydroxymethyl. Water, on the other hand, heterolytically oxidizes hydrogen to produce hydronium ions and electrons that reduce oxygen. These findings suggest that reactions of solvent molecules at solid-liquid interfaces can generate redox mediators in situ and provide opportunities to substantially increase rates and selectivities for catalytic reactions.
Effect of Structural Ordering on the Charge Storage Mechanism of p-Type Organic Electrode Materials
ACS AMI
 Brian M. Peterson, Cara N. Gannett, Luis Melecio-Zambrano, Brett P. Fors, and Héctor Abruña
Brian M. Peterson, Cara N. Gannett, Luis Melecio-Zambrano, Brett P. Fors, and Héctor Abruña
Effect of Structural Ordering on the Charge Storage Mechanism of p-Type Organic Electrode Materials
Understanding the properties that govern the kinetics of charge storage will enable informed design strategies and improve the rate performance of future battery materials. Herein, we study the effects of structural ordering in organic electrode materials on their charge storage mechanisms. A redox active unit, N,N′-diphenyl-phenazine, was incorporated into three materials which exhibited varying degrees of ordering. From cyclic voltammetry analysis, the crystalline small molecule exhibited diffusion-limited behavior, likely arising from structural rearrangements that occur during charge/discharge. Conversely, a branched polymer network displayed surface-controlled kinetics, attributed to the amorphous structure which enabled fast ionic transport throughout charge/discharge, unimpeded by sluggish structural rearrangements. These results suggest a method for designing new materials for battery electrodes with battery-like energy densities and pseudocapacitor-like rate capabilities.
Organic Electrode Materials for Fast-Rate, High-Power Battery Applications
Materials Reports: Energy
Cara N. Gannett, Luis Melecio-Zambrano, Monica Theibault, Brian M. Peterson, Brett P. Fors, Héctor D. Abruña
Organic Electrode Materials for Fast-Rate, High-Power Battery Applications
The development of new battery materials with fast charging/discharging capabilities is necessary to meet the growing demands of modern technologies. While counter ion transport in inorganic materials (generally by de/intercalation) currently limits charge/discharge rates in lithium-ion batteries, the weak intermolecular forces in organic materials result in flexible, spacious structures that offer improved ion transport capabilities. Herein, we present the principles which enable fast rate capabilities in organic electrode materials, accompanied by specific literature examples illustrating exceptional rate performances. We discuss approaches to material design which support electron and/or ion transport and the limitations associated with each. This review aims to highlight the unique characteristics of organic materials as high-power density electrodes and inspire continued work in the field.
Performance optimization and fast rate capabilities of novel polymer cathode materials through balanced electronic and ionic transport
Journal of Materials Chemistry A
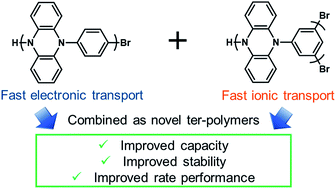
Cara N. Gannett, Brian M. Peterson, Luis Melecio-Zambrano, Colleen Q. Trainor, Brett P. Fors and Héctor D. Abruña
Performance optimization and fast rate capabilities of novel polymer cathode materials through balanced electronic and ionic transport
The increasing demands for high-power electrical energy storage technologies require the development of new electrode materials and architectures with fast ion and electron transport. Herein, we report a new family of ter-polymers as battery cathode materials which exhibit significantly improved performance over their parent co-polymers: poly(phenylene-phenazine), and poly(1,3,5-phenylene-phenazine). The high electronic conductivity of poly(phenylene-phenazine) and fast ionic transport of poly(1,3,5-phenylene-phenazine) are combined in this series of ter-polymers, exhibiting improved battery performances which are especially apparent at high rates. The optimized ter-polymer delivers 180 mA h g−1 when discharged at 16 A g−1, demonstrating the effectiveness of balancing ionic and electronic transport properties.
Carbonyl Desaturation: Where Does Catalysis Stand?
ACS Catalysis

Samer Gnaim, Julien C. Vantourout, Fabien Serpier, Pierre-Georges Echeverria, and Phil. S. Baran
Carbonyl Desaturation: Where Does Catalysis Stand?
There is a strong parallel between simple alcohol oxidation and carbonyl desaturation from both strategic and tactical vantage points. As they both seek to extract hydrogen from an organic substrate, they are deceptively simple looking transformations that have been addressed over the past 70+ years through stoichiometric means. The past decade has seen an intensifying level of interest in rendering both of these simple reactions catalytic. In this Perspective, recent advances from the past 5 years are highlighted featuring both transition-metal-catalyzed and metal-free approaches to carbonyl desaturation. Through a historical overview and a detailed look at each of these new developments, we seek to address the question of in what context a catalytic strategy emerges as ideal.Electroreductive Olefin–Ketone Coupling
JACS
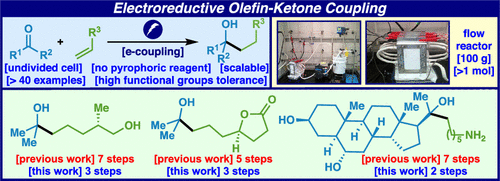
Pengfei Hu, Byron K. Peters, Christian A. Malapit, Julien C. Vantourout, Pan Wang, Jinjun Li, Lucas Mele, Pierre-Georges Echeverria, Shelley D. Minteer, and Phil S. Baran
Electroreductive Olefin–Ketone Coupling
A user-friendly approach is presented to sidestep the venerable Grignard addition to unactivated ketones to access tertiary alcohols by reversing the polarity of the disconnection. In this work a ketone instead acts as a nucleophile when adding to simple unactivated olefins to accomplish the same overall transformation. The scope of this coupling is broad as enabled using an electrochemical approach, and the reaction is scalable, chemoselective, and requires no precaution to exclude air or water. Multiple applications demonstrate the simplifying nature of the reaction on multistep synthesis, and mechanistic studies point to an intuitive mechanism reminiscent of other chemical reductants such as SmI2 (which cannot accomplish the same reaction).
Biphasic Bioelectrocatalytic Synthesis of Chiral β-Hydroxy Nitriles
JACS
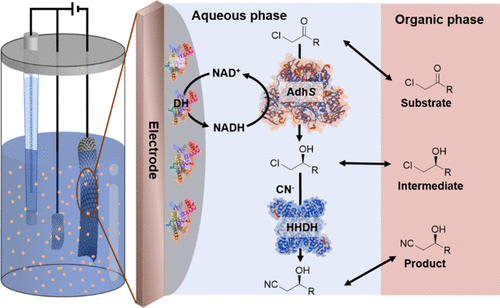
Fangyuan Dong, Hui Chen, Christian A. Malapit, Matthew B. Prater, Min Li, Mengwei Yuan, Koun Lim, and Shelley D. Minteer
Biphasic Bioelectrocatalytic Synthesis of Chiral β-Hydroxy Nitriles
Two obstacles limit the application of oxidoreductase-based asymmetric synthesis. One is the consumption of high stoichiometric amounts of reduced cofactor. The other is the low solubility of organic substrates, intermediates, and products in the aqueous phase. In order to address these two obstacles to oxidoreductase-based asymmetric synthesis, a biphasic bioelectrocatalytic system was constructed and applied. In this study, the preparation of chiral β-hydroxy nitriles catalyzed by alcohol dehydrogenase (AdhS) and halohydrin dehalogenase (HHDH) was investigated as a model bioelectrosynthesis, since they are high-value intermediates in statin synthesis. Diaphorase (DH) was immobilized by a cobaltocene-modified poly(allylamine) redox polymer on the electrode surface (DH/Cc-PAA bioelectrode) to achieve effective bioelectrocatalytic NADH regeneration. Since AdhS is a NAD-dependent dehydrogenase, the diaphorase-modified biocathode was used to regenerate NADH to support the conversion from ethyl 4-chloroacetoacetate (COBE) to ethyl (S)-4-chloro-3-hydroxybutanoate ((S)-CHBE) catalyzed by AdhS. The addition of methyl tert-butyl ether (MTBE) as an organic phase not only increased the uploading of COBE but also prevented the spontaneous hydrolysis of COBE, extended the lifetime of DH/Cc-PAA bioelectrode, and increased the Faradaic efficiency and the concentration of generated (R)-ethyl-4-cyano-3-hydroxybutyrate ((R)-CHCN). After 10 h of reaction, the highest concentration of (R)-CHCN in the biphasic bioelectrocatalytic system was 25.5 mM with 81.2% enantiomeric excess (eep). The conversion ratio of COBE achieved 85%, which was 8.8 times higher than that achieved with the single-phase system. Besides COBE, two other substrates with aromatic ring structures were also used in this biphasic bioelectrocatalytic system to prepare the corresponding chiral β-hydroxy nitriles. The results indicate that the biphasic bioelectrocatalytic system has the potential to produce a variety of β-hydroxy nitriles with different structures.
Selective Electroenzymatic Oxyfunctionalization by Alkane Monooxygenase in a Biofuel Cell
Angewandte Chemie
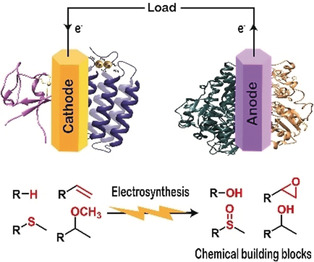
Mengwei Yuan, Sofiene Abdellaoui, Hui Chen, Matthew J. Kummer, Christian A. Malapit, Chun You, Shelley D. Minteer
Selective Electroenzymatic Oxyfunctionalization by Alkane Monooxygenase in a Biofuel Cell
Aliphatic synthetic intermediates with high added value are generally produced from alkane sources (e.g., petroleum) by inert carbon–hydrogen (C−H) bond activation using classical chemical methods (i.e. high temperature, rare metals). As an alternative approach for these reactions, alkane monooxygenase from Pseudomonas putida (alkB) is able to catalyze the difficult terminal oxyfunctionalization of alkanes selectively and under mild conditions. Herein, we report an electrosynthetic system using an alkB biocathode which produces alcohols, epoxides, and sulfoxides through bioelectrochemical hydroxylation, epoxidation, sulfoxidation, and demethylation. The capacity of the alkB binding pocket to protect internal functional groups is also demonstrated. By coupling our alkB biocathode with a hydrogenase bioanode and using H2 as a clean fuel source, we have developed and characterized a series of enzymatic fuel cells capable of oxyfunctionalization while simultaneously producing electricity.
Electrochemical Reduction of [Ni(Mebpy)3]2+: Elucidation of the Redox Mechanism by Cyclic Voltammetry and Steady‐State Voltammetry in Low Ionic Strength Solutions
ChemElectroChem
Koushik Barman, Martin A. Edwards, David P. Hickey, Christopher Sandford, Yinghua Qiu, Rui Gao, Shelley D. Minteer, Henry S. White
Electrochemical Reduction of [Ni(Mebpy)3]2+: Elucidation of the Redox Mechanism by Cyclic Voltammetry and Steady‐State Voltammetry in Low Ionic Strength Solutions
Bipyridine complexes of Ni are used as catalysts in a variety of reductive transformations. Here, the electroreduction of [Ni(Mebpy)3]2+ (Mebpy=4,4′‐dimethyl‐2,2′‐bipyridine) in dimethylformamide is reported, with the aim of determining the redox mechanism and oxidation states of products formed under well‐controlled electrochemical conditions. Results from cyclic voltammetry, steady‐state voltammetry (SSV) and chronoamperometry demonstrate that [Ni(Mebpy)3]2+ undergoes two sequential 1e reductions at closely separated potentials (E10’=−1.06±0.01 V and E20’=−1.15±0.01 V vs Ag/AgCl (3.4 M KCl)). Homogeneous comproportionation to generate [Ni(Mebpy)3]+ is demonstrated in SSV experiments in low ionic strength solutions. The comproportionation rate constant is determined to be >106 M−1 s−1, consistent with rapid outer‐sphere electron transfer. Consequentially, on voltammetric time scales, the 2e reduction of [Ni(Mebpy)3]2+ results in formation of [Ni(Mebpy)3]+ as the predominant species released into bulk solution. We also demonstrate that [Ni(Mebpy)3]0 slowly loses a Mebpy ligand (∼10 s−1).
Ionic Liquid Stabilized 2,2,6,6-Tetramethylpiperidine 1-Oxyl Catalysis for Alcohol Oxidation
ACS Sustainable Chemistry & Engineering
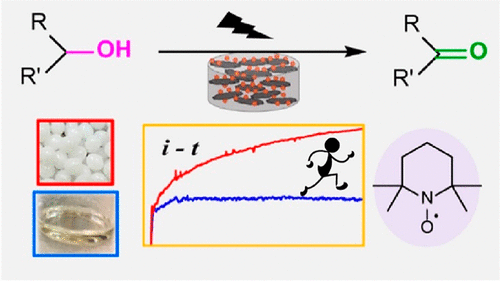
Min Li, Kevin Klunder, Emmy Blumenthal, Matthew B. Prater, Jack Lee, John E. Matthiesen, and Shelley D. Minteer
Ionic Liquid Stabilized 2,2,6,6-Tetramethylpiperidine 1-Oxyl Catalysis for Alcohol Oxidation
N-Oxyl reagents, particularly 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), have been extensively used for alcohol oxidations. While TEMPO-mediated oxidations are kinetically and thermodynamically favorable in high-pH electrolytes, base-induced degradation often results in significant loss of catalytic activity. Herein, we demonstrate enhanced alkaline stability of a TEMPO derivative in ionic liquids (ILs). By incorporating TEMPO in an imidazole-anchored IL, no loss of current was observed at pH 10.0 after 2.0 h during the oxidation of butanol and glycerol, while TEMPO in polycaprolactone (PCL), a patternable binder material, degraded 58.5% and 67.1%, respectively. The stability enhancement was further demonstrated by analyzing the conversion of glycerol in an 800 μL electrochemical cell using bulk chemical analysis techniques. Successive cycles of glycerol oxidation indicated 14-fold stability enhancement by applying IL in a TEMPO electrode composite in comparison to PCL. The strategy demonstrated here provides an opportunity to prepare catalytic systems with enhanced stability. Further, this method provides the ability to convert what are typically homogeneous catalysts to heterogeneous systems.
Bioelectrocatalytic Conversion from N2 to Chiral Amino Acids in a H2/α-Keto Acid Enzymatic Fuel Cell
JACS
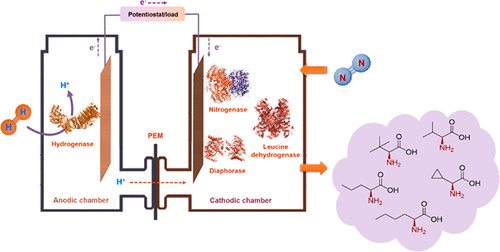
Hui Chen, Matthew B. Prater, Rong Cai, Fangyuan Dong, Hsiaonung Chen, and Shelley D. Minteer
Bioelectrocatalytic Conversion from N2 to Chiral Amino Acids in a H2/α-Keto Acid Enzymatic Fuel Cell
Enzymatic electrosynthesis is a promising approach to produce useful chemicals with the requirement of external electrical energy input. Enzymatic fuel cells (EFCs) are devices to convert chemical energy to electrical energy via the oxidation of fuel at the anode and usually the reduction of oxygen or peroxide at the cathode. The integration of enzymatic electrosynthesis with EFC architectures can simultaneously result in self-powered enzymatic electrosynthesis with more valuable usage of electrons to produce high-value-added chemicals. In this study, a H2/α-keto acid EFC was developed for the conversion from chemically inert nitrogen gas to chiral amino acids, powered by H2 oxidation. A highly efficient cathodic reaction cascade was first designed and constructed. Powered by an applied voltage, the cathode supplied enough reducing equivalents to support the NH3 production and NADH recycling catalyzed by nitrogenase and diaphorase. The produced NH3 and NADH were reacted in situ with leucine dehydrogenase (LeuDH) to generate l-norleucine with 2-ketohexanoic acid as the NH3 acceptor. A 92% NH3 conversion ratio and 87.1% Faradaic efficiency were achieved. On this basis, a H2-powered fuel cell with hyper-thermostable hydrogenase (SHI) as the anodic catalyst was combined with the cathodic reaction cascade to form the H2/α-keto acid EFC. After 10 h of reaction, the concentration of l-norleucine achieved 0.36 mM with >99% enantiomeric excess and 82% Faradaic efficiency. From the broad substrate scope and the high enzymatic enantioselectivity of LeuDH, the H2/α-keto acid EFC is an energy-efficient alternative to electrochemically produce chiral amino acids for biotechnology applications.
The progress and outlook of bioelectrocatalysis for the production of chemicals, fuels and materials
Nature Catalysis
Hui Chen, Fangyuan Dong & Shelley D. Minteer
The progress and outlook of bioelectrocatalysis for the production of chemicals, fuels and materials
Bioelectrocatalysis is a green, sustainable, efficient method to produce value-added chemicals, clean biofuels and degradable materials. As an alternative approach to modern biomanufacturing technology, bioelectrocatalysis fully combines the merits of both biocatalysis and electrocatalysis to realize the green, efficient production of target products from electricity. Here, we review the development status of bioelectrocatalysis, discussing the current challenges and looking toward future development directions. First, we detail the structure, function and modification methods of bioelectrocatalysis. Next, we describe the mechanism of electron transfer, including mediated electron transfer and directed electron transfer. Third, we discuss the impact of the electrode on bioelectrocatalysis. Then we analyse and summarize the application of bioelectrocatalysis methods in the production of chemicals, biofuels and materials. Finally, we detail future developments and perspectives on bioelectrocatalysis for electrosynthesis.
A Survival Guide for the “Electro-curious”
Accounts of Chemical Research
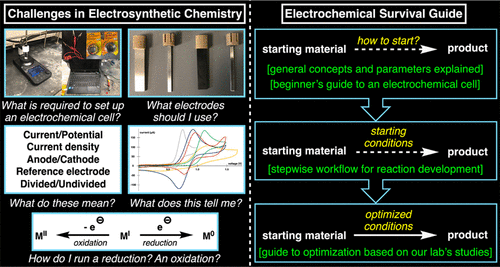
Cian Kingston, Maximilian D. Palkowitz, Yusuke Takahira, Julien C. Vantourout, Byron K. Peters, Yu Kawamata, and Phil S. Baran
A Survival Guide for the “Electro-curious”
The appeal and promise of synthetic organic electrochemistry have been appreciated over the past century. In terms of redox chemistry, which is frequently encountered when forging new bonds, it is difficult to conceive of a more economical way to add or remove electrons than electrochemistry. Indeed, many of the largest industrial synthetic chemical processes are achieved in a practical way using electrons as a reagent. Why then, after so many years of the documented benefits of electrochemistry, is it not more widely embraced by mainstream practitioners? Erroneous perceptions that electrochemistry is a “black box” combined with a lack of intuitive and inexpensive standardized equipment likely contributed to this stagnation in interest within the synthetic organic community. This barrier to entry is magnified by the fact that many redox processes can already be accomplished using simple chemical reagents even if they are less atom-economic. Time has proven that sustainability and economics are not strong enough driving forces for the adoption of electrochemical techniques within the broader community. Indeed, like many synthetic organic chemists that have dabbled in this age-old technique, our first foray into this area was not by choice but rather through sheer necessity.
The unique reactivity benefits of this old redox-modulating technique must therefore be highlighted and leveraged in order to draw organic chemists into the field. Enabling new bonds to be forged with higher levels of chemo- and regioselectivity will likely accomplish this goal. In doing so, it is envisioned that widespread adoption of electrochemistry will go beyond supplanting unsustainable reagents in mundane redox reactions to the development of exciting reactivity paradigms that enable heretofore unimagined retrosynthetic pathways. Whereas the rigorous physical organic chemical principles of electroorganic synthesis have been reviewed elsewhere, it is often the case that such summaries leave out the pragmatic aspects of designing, optimizing, and scaling up preparative electrochemical reactions. Taken together, the task of setting up an electrochemical reaction, much less inventing a new one, can be vexing for even seasoned organic chemists. This Account therefore features a unique format that focuses on addressing this exact issue within the context of our own studies. The graphically rich presentation style pinpoints basic concepts, typical challenges, and key insights for those “electro-curious” chemists who seek to rapidly explore the power of electrochemistry in their research.
Nitrogenase Bioelectrochemistry for Synthesis Applications
Accounts of Chemical Research
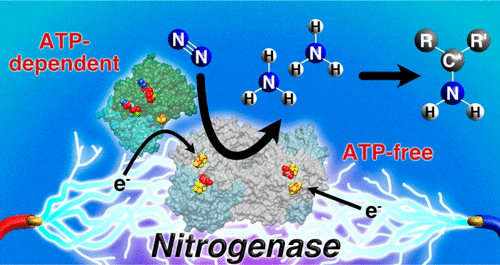
Ross D. Milton and Shelley D. Minteer
Nitrogenase Bioelectrochemistry for Synthesis Applications
The fixation of atmospheric dinitrogen to ammonia by industrial technologies (such as the Haber Bosch process) has revolutionized humankind. In contrast to industrial technologies, a single enzyme is known for its ability to reduce or “fix” dinitrogen: nitrogenase. Nitrogenase is a complex oxidoreductase enzymatic system that includes a catalytic protein (where dinitrogen is reduced) and an electron-transferring reductase protein (termed the Fe protein) that delivers the electrons necessary for dinitrogen fixation. The catalytic protein most commonly contains a FeMo cofactor (called the MoFe protein), but it can also contain a VFe or FeFe cofactor. Besides their ability to fix dinitrogen to ammonia, these nitrogenases can also reduce substrates such as carbon dioxide to formate. Interestingly, the VFE nitrogenase can also form carbon–carbon bonds. The vast majority of research surrounding nitrogenase employs the Fe protein to transfer electrons, which is also associated with the rate-limiting step of nitrogenase catalysis and also requires the hydrolysis of adenosine triphosphate. Thus, there is significant interest in artificially transferring electrons to the catalytic nitrogenase proteins. In this Account, we review nitrogenase electrocatalysis whereby electrons are delivered to nitrogenase from electrodes. We first describe the use of an electron mediator (cobaltocene) to transfer electrons from electrodes to the MoFe protein. The reduction of protons to molecular hydrogen was realized, in addition to azide and nitrite reduction to ammonia. Bypassing the rate-limiting step within the Fe protein, we also describe how this approach was used to interrogate the rate-limiting step of the MoFe protein: metal-hydride protonolysis at the FeMo-co. This Account next reviews the use of cobaltocene to mediate electron transfer to the VFe protein, where the reduction of carbon dioxide and the formation of carbon–carbon bonds (yielding the formation of ethene and propene) was realized. This approach also found success in mediating electron transfer to the FeFe catalytic protein, which exhibited improved carbon dioxide reduction in comparison to the MoFe protein. In the final example of mediated electron transfer to the catalytic protein, this Account also reviews recent work where the coupling of infrared spectroscopy with electrochemistry enabled the potential-dependent binding of carbon monoxide to the FeMo-co to be studied. As an alternative to mediated electron transfer, recent work that has sought to transfer electrons to the catalytic proteins in the absence of electron mediators (by direct electron transfer) is also reviewed. This approach has subsequently enabled a thermodynamic landscape to be proposed for the cofactors of the catalytic proteins. Finally, this Account also describes nitrogenase electrocatalysis whereby electrons are first transferred from an electrode to the Fe protein, before being transferred to the MoFe protein alongside the hydrolysis of adenosine triphosphate. In this way, increased quantities of ammonia can be electrocatalytically produced from dinitrogen fixation. We discuss how this has led to the further upgrade of electrocatalytically produced ammonia, in combination with additional enzymes (diaphorase, alanine dehydrogenase, and transaminase), to selective production of chiral amine intermediates for pharmaceuticals. This Account concludes by discussing current and future research challenges in the field of electrocatalytic nitrogen fixation by nitrogenase.
Polycaprolactone-enabled sealing and carbon composite electrode integration into electrochemical microfluidics
Lab on a Chip
Kevin J. Klunder, Kaylee M. Clark, Cynthia McCord, Kathleen E. Berg, Shelley D. Minteer, Charles S. Henry
Polycaprolactone-enabled sealing and carbon composite electrode integration into electrochemical microfluidics
Combining electrochemistry with microfluidics is attractive for a wide array of applications including multiplexing, automation, and high-throughput screening. Electrochemical instrumentation also has the advantage of being low-cost and can enable high analyte sensitivity. For many electrochemical microfluidic applications, carbon electrodes are more desirable than noble metals because they are resistant to fouling, have high activity, and large electrochemical solvent windows. At present, fabrication of electrochemical microfluidic devices bearing integrated carbon electrodes remains a challenge. Here, a new system for integrating polycaprolactone (PCL) and carbon composite electrodes into microfluidics is presented. The PCL : carbon composites have excellent electrochemical activity towards a wide range of analytes as well as high electrical conductivity (∼1000 S m−1). The new system utilizes a laser cutter for fast, simple fabrication of microfluidics using PCL as a bonding layer. As a proof-of-concept application, oil-in-water and water-in-oil droplets are electrochemically analysed. Small-scale electrochemical organic synthesis for TEMPO mediated alcohol oxidation is also demonstrated.
Mechanistic Studies into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization
JACS
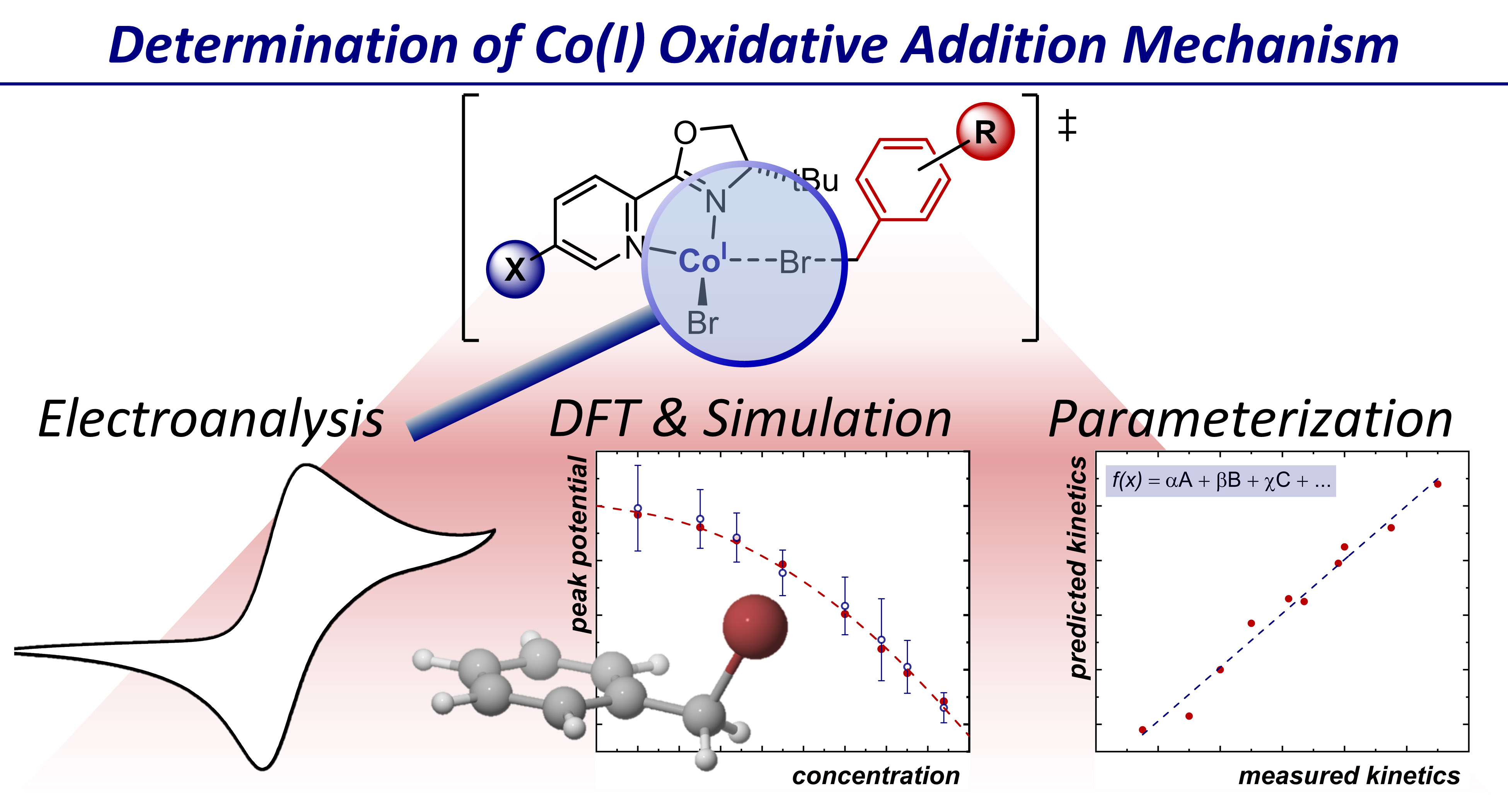
Christopher Sandford, Lydia R. Fries, Tyler E. Ball, Shelley D. Minteer, and Matthew S Sigman
Mechanistic Studies into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization
The oxidative addition of organic electrophiles into electrochemically generated Co(I) complexes has been widely utilized as a strategy to produce carbon-centered radicals when cobalt is ligated by a polydentate ligand. Changing to a bidentate ligand provides the opportunity to access discrete Co(III)ñC bonded complexes for alternative reactivity, but knowledge of how ligand and/or substrate structures affect catalytic steps is pivotal to reaction design and catalyst optimization. In this vein, experimental studies which can determine the exact nature of elementary organometallic steps remain limited, especially for single-electron oxidative addition pathways. Herein, we utilize cyclic voltammetry combined with simulations to obtain kinetic and thermodynamic properties of the two-step, halogen-atom abstraction mechanism, validated by analyzing kinetic isotope and substituent effects. Complex Hammett relationships could be disentangled to allow understanding of individual effects on activation energy barriers and equilibrium constants, and DFT-derived parameters used to build predictive statistical models for rates of new ligand/substrate combinations.Hindered dialkyl ether synthesis with electrogenerated carbocations
Nature
Jinbao Xiang, Ming Shang, Yu Kawamata, Helena Lundberg, Solomon H. Reisberg, Miao Chen, Pavel Mykhailiuk, Gregory Beutner, Michael R. Collins, Alyn Davies, Matthew Del Bel, Gary M. Gallego, Jillian E. Spangler, Jeremy Starr, Shouliang Yang, Donna G. Blackmond & Phil S. Baran
Hindered dialkyl ether synthesis with electrogenerated carbocations
Hindered ethers are of high value for various applications; however, they remain an underexplored area of chemical space because they are difficult to synthesize via conventional reactions’–1,–2. Such motifs are highly coveted in medicinal chemistry, because extensive substitution about the ether bond prevents unwanted metabolic processes that can lead to rapid degradation in vivo. Here we report a simple route towards the synthesis of hindered ethers, in which electrochemical oxidation is used to liberate high-energy carbocations from simple carboxylic acids. These reactive carbocation intermediates, which are generated with low electrochemical potentials, capture an alcohol donor under non-acidic conditions; this enables the formation of a range of ethers (more than 80 have been prepared here) that would otherwise be difficult to access. The carbocations can also be intercepted by simple nucleophiles, leading to the formation of hindered alcohols and even alkyl fluorides. This method was evaluated for its ability to circumvent the synthetic bottlenecks encountered in the preparation of 12 chemical scaffolds, leading to higher yields of the required products, in addition to substantial reductions in the number of steps and the amount of labour required to prepare them. The use of molecular probes and the results of kinetic studies support the proposed mechanism and the role of additives under the conditions examined. The reaction manifold that we report here demonstrates the power of electrochemistry to access highly reactive intermediates under mild conditions and, in turn, the substantial improvements in efficiency that can be achieved with these otherwise-inaccessible intermediates.Efficient NADH Regeneration by a Redox Polymer-Immobilized Enzymatic System
ACS Catalysis
Efficient NADH Regeneration by a Redox Polymer-Immobilized Enzymatic System
Given the high costs and stoichiometric amounts of reduced nicotinamide adenine dinucleotide (NADH) required by the many oxidoreductases used for organic synthesis and the pharmaceutical industry, there is a need for the efficient reductive regeneration of NADH from its oxidized form, NAD+. Bioelectrocatalytic methods for NADH regeneration involving diaphorase and a redox mediator have shown promise; however, strong reductive mediators needed for this system are scarce, generally unstable, and require downstream separation. The immobilization of diaphorase in cobaltocene-modified poly(allylamine) redox polymer is presented which is capable of producing bioactive 1,4-NADH with yields between 97% and 100%, faradaic efficiencies between 78% and 99%, and turnover frequencies between 2091 h–1 and 3680 h–1 over a range of temperatures spanning 20 to 60 °C. By using this system, methanol and propanol production by an NADH-dependent alcohol dehydrogenase were enhanced 7.1- and 5.2-fold, respectively, compared with a negative control. Finally, the efficiency of this approach coupled with its high operational stability (91% of the maximum activity after five experimental cycles) renders it among the most promising means of NADH regeneration yet developed..A synthetic chemist's guide to electroanalytical tools for studying reaction mechanisms
Chemical Science
Christopher Sandford, Martin A. Edwards, Kevin J. Klunder, David P. Hickey, Min Li, Koushik Barman, Matthew S. Sigman, Henry S. White, and Shelley D. Minteer
A synthetic chemist's guide to electroanalytical tools for studying reaction mechanisms
Monitoring reactive intermediates can provide vital information in the study of synthetic reaction mechanisms, enabling the design of new catalysts and methods. Many synthetic transformations are centred on the alteration of oxidation states, but these redox processes frequently pass through intermediates with short life-times, making their study challenging. A variety of electroanalytical tools can be utilised to investigate these redox-active intermediates: from voltammetry to in situ spectroelectrochemistry and scanning electrochemical microscopy. This perspective provides an overview of these tools, with examples of both electrochemically-initiated processes and monitoring redox-active intermediates formed chemically in solution. The article is designed to introduce synthetic organic and organometallic chemists to electroanalytical techniques and their use in probing key mechanistic questions.Phthalocyanines as a π–π Adsorption Strategy to Immobilize Catalyst on Carbon for Electrochemical Synthesis
Synlett
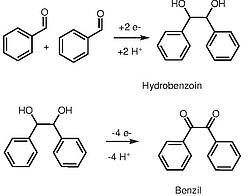
Kevin Klunder, Ashley Cass, Scott Anderson, Shelley Minteer
Phthalocyanines as a π–π Adsorption Strategy to Immobilize Catalyst on Carbon for Electrochemical Synthesis
In most electrochemical syntheses, reactions are happening at or near the electrode surface. For catalyzed reactions, ideally, the electrode surface would solely contain the catalyst, which then simplifies purification and lowers the amount of catalyst needed. Here, a new strategy involving phthalocyanines (Pc) to immobilize catalysts onto carbon electrode surfaces is presented. The large π structure of the Pc enables adsorption to the sp2-structure of graphitic carbon. TEMPO-modified Pc were chosen as a proof of concept to test the new immobilization strategy. It was found that the TEMPO-Pc derivatives functioned similarly or better than the widely used pyrene adsorption method. Interestingly, the new TEMPO-Pc catalyst appears to facilitate a cascade reaction involving both the anode and the cathode. The first step is the generation of an aryl aldehyde (anode) followed by the reduction of the aryl aldehyde in a pinacol-type coupling reaction at the cathode. The last step is the oxidation of a hydrobenzoin to create benzil. This work demonstrates the unique ability of electrochemistry and bifunctional catalysts to enable multistep chemical transformations, performing both reductive and oxidative transformations in one pot.
Upgraded Bioelectrocatalytic N2 Fixation: From N2 to Chiral Amine Intermediates
JACS
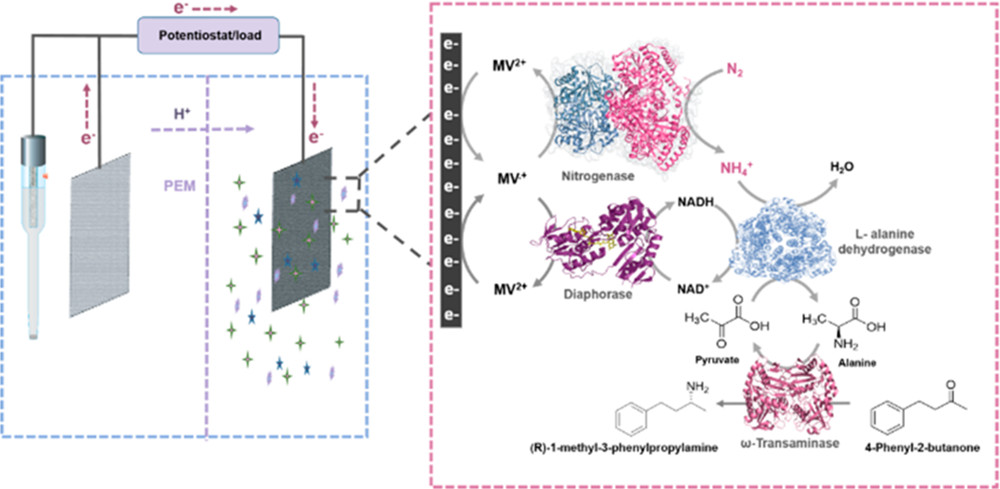
Upgraded Bioelectrocatalytic N2 Fixation: From N2 to Chiral Amine Intermediates
Enantiomerically pure chiral amines are of increasing value in the preparation of bioactive compounds, pharmaceuticals, and agrochemicals. ω-Transaminase (ω-TA) is an ideal catalyst for asymmetric amination because of its excellent enantioselectivity and wide substrate scope. To shift the equilibrium of reactions catalyzed by ω-TA to the side of the amine product, an upgraded N2 fixation system based on bioelectrocatalysis was developed to realize the conversion from N2to chiral amine intermediates. The produced NH3 was in situ reacted with l-alanine dehydrogenase to generate alanine with NADH as a coenzyme. ω-TA transferred the amino group from alanine to ketone substrates and finally produced the desired chiral amine intermediates. The cathode of the upgraded N2 fixation system supplied enough reducing power to synchronously realize the regeneration of reduced methyl viologen (MV•+) and NADH for the nitrogenase and l-alanine dehydrogenase. The coproduct, pyruvate, was consumed by l-alanine dehydrogenase to regenerate alanine and push the equilibrium to the side of amine. After 10 h of reaction, the concentration of 1-methyl-3-phenylpropylamine achieved 0.54 mM with the 27.6% highest faradaic efficiency and >99% enantiomeric excess (eep). Because of the wide substrate scope and excellent enantioselectivity of ω-TA, the upgraded N2 fixation system has great potential to produce a variety of chiral amine intermediates for pharmaceuticals and other applications.Electrochemical C(sp3)–H Fluorination
Synlett
Electrochemical C(sp3)–H Fluorination
A simple and robust method for electrochemical alkyl C–H fluorination is presented. Using a simple nitrate additive, a widely available fluorine source (Selectfluor), and carbon-based electrodes, a wide variety of activated and unactivated C–H bonds are converted into their C–F congeners. The scalability of the reaction is also demonstrated with a 100 gram preparation of fluorovaline.Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications
JACS
Yu Kawamata, Julien Christian Vantourout, David P. Hickey, Peng Bai, Longrui Chen, Qinglong Hou, Wenhua Qiao, Koushik Barman, Martin A. Edwards, Alberto F. Garrido-Castro, Justine N. deGruyter, Hugh Nakamura, Kyle W. Knouse, Chuanguang Qin, Khalyd J. Clay, Denghui Bao, Chao Li, Jeremy T. Starr, Carmen Garcia-Irizarry, Neal Sach, Henry S. White, Matthew Neurock, Shelley D. Minteer, Phil S. Baran
Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications
C–N cross-coupling is one of the most valuable and widespread transformations in organic synthesis. Largely domi-nated by Pd- and Cu-based catalytic systems, it has proven to be a staple transformation for those in both academia and industry. The current study presents the development and mechanistic understanding of an electrochemically driven, Ni-catalyzed method for achieving this reaction of high strategic importance. Through a series of electro-chemical, computational, kinetic, and empirical experiments the key mechanistic features of this reaction have been unraveled, leading to a second generation set of conditions that is applicable to a broad range of aryl halides and amine nucleophiles, including complex examples on oligopeptides, medicinally-relevant heterocycles, natu-ral products, and sugars. Full disclosure of the current limitations as well as procedures for both batch and flow scale-ups (100 gram) are also described.
Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-ion Battery Chemistry
Science
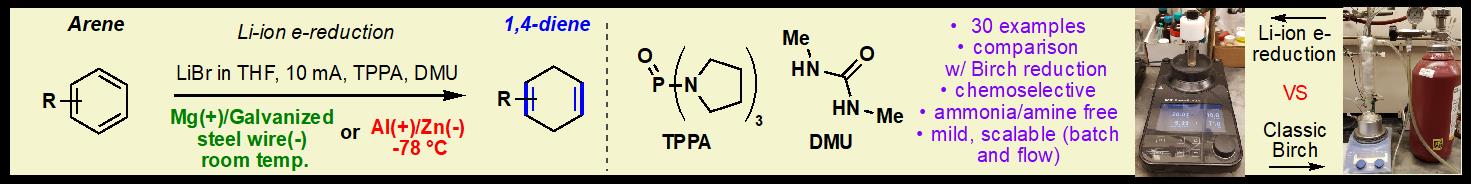
Byron K. Peters, Kevin X. Rodriguez, Solomon H. Reisberg, Sebastian B. Beil, David P. Hickey, Yu Kawamata, Michael Collins, Jeremy Starr, Longrui Chen, Sagar Udyavara, Kevin Klunder, Timothy J. Gorey, Scott L. Anderson, Matthew Neurock, Shelley D. Minteer, Phil S. Baran
Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-ion Battery Chemistry
Reductive electrosynthesis has faced long-standing challenges in applications to complex organic substrates at scale. Here, we show how decades of research in lithium-ion battery materials, electrolytes, and additives can serve as an inspiration for achieving practically scalable reductive electrosynthetic conditions for the Birch reduction. Specifically, we demonstrate that using a sacrificial anode material (magnesium or aluminum), combined with a cheap, nontoxic, and water-soluble proton source (dimethylurea), and an overcharge protectant inspired by battery technology [tris(pyrrolidino)phosphoramide] can allow for multigram-scale synthesis of pharmaceutically relevant building blocks. We show how these conditions have a very high level of functional-group tolerance relative to classical electrochemical and chemical dissolving-metal reductions. Finally, we demonstrate that the same electrochemical conditions can be applied to other dissolving metal–type reductive transformations, including McMurry couplings, reductive ketone deoxygenations, and epoxide openings.
Investigating the Role of Ligand Electronics on Stabilizing Electrocatalytically Relevant Low-Valent Co(I) Intermediates
JACS
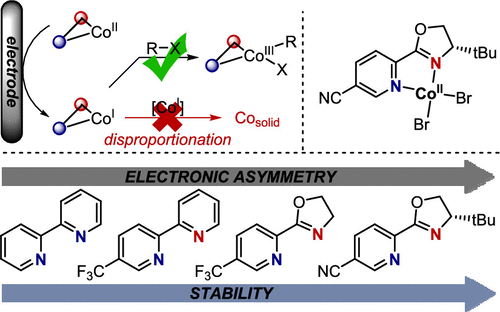
David P. Hickey, Christopher Sandford, Zayn Rhodes, Tobias Gensch, Lydia R. Fries, Matthew S. Sigman, and Shelley D. Minteer
Investigating the Role of Ligand Electronics on Stabilizing Electrocatalytically Relevant Low-Valent Co(I) Intermediates
Cobalt complexes have shown great promise as electrocatalysts in applications ranging from hydrogen evolution to C–H functionalization. However, the use of such complexes often requires polydentate, bulky ligands to stabilize the catalytically active Co(I) oxidation state from deleterious disproportionation reactions to enable the desired reactivity. Herein, we describe the use of bidentate electronically asymmetric ligands as an alternative approach to stabilizing transient Co(I) species. Using disproportionation rates of electrochemically generated Co(I) complexes as a model for stability, we measured the relative stability of complexes prepared with a series of N,N-bidentate ligands. While the stability of Co(I)Cl complexes demonstrates a correlation with experimentally measured thermodynamic properties, consistent with an outer-sphere electron transfer process, the set of ligated Co(I)Br complexes evaluated was found to be preferentially stabilized by electronically asymmetric ligands, demonstrating an alternative disproportionation mechanism. These results allow a greater understanding of the fundamental processes involved in the disproportionation of organometallic complexes and have allowed the identification of cobalt complexes that show promise for the development of novel electrocatalytic reactions.